Clear Sky Science · fr
Modélisation multi‑échelle des zones GPAl‑Li dans les alliages Al‑Li à partir des principes premiers
Pourquoi les métaux légers comptent
Des fusées et réservoirs de carburant aux avions de nouvelle génération, les concepteurs recherchent des métaux à la fois légers et résistants. Les alliages aluminium‑lithium sont des candidats de choix car une faible quantité de lithium rend l’aluminium plus léger et plus rigide. Pourtant, la résistance de ces alliages provient de petits amas d’atomes difficiles à détecter qui se forment à l’intérieur du métal lors du traitement thermique. Cet article s’attaque à un mystère de longue date concernant l’un de ces amas, l’éphémère zone GPAl–Li, et montre comment elle s’intègre dans la chaîne de transformations qui confèrent à l’alliage ses propriétés remarquables.
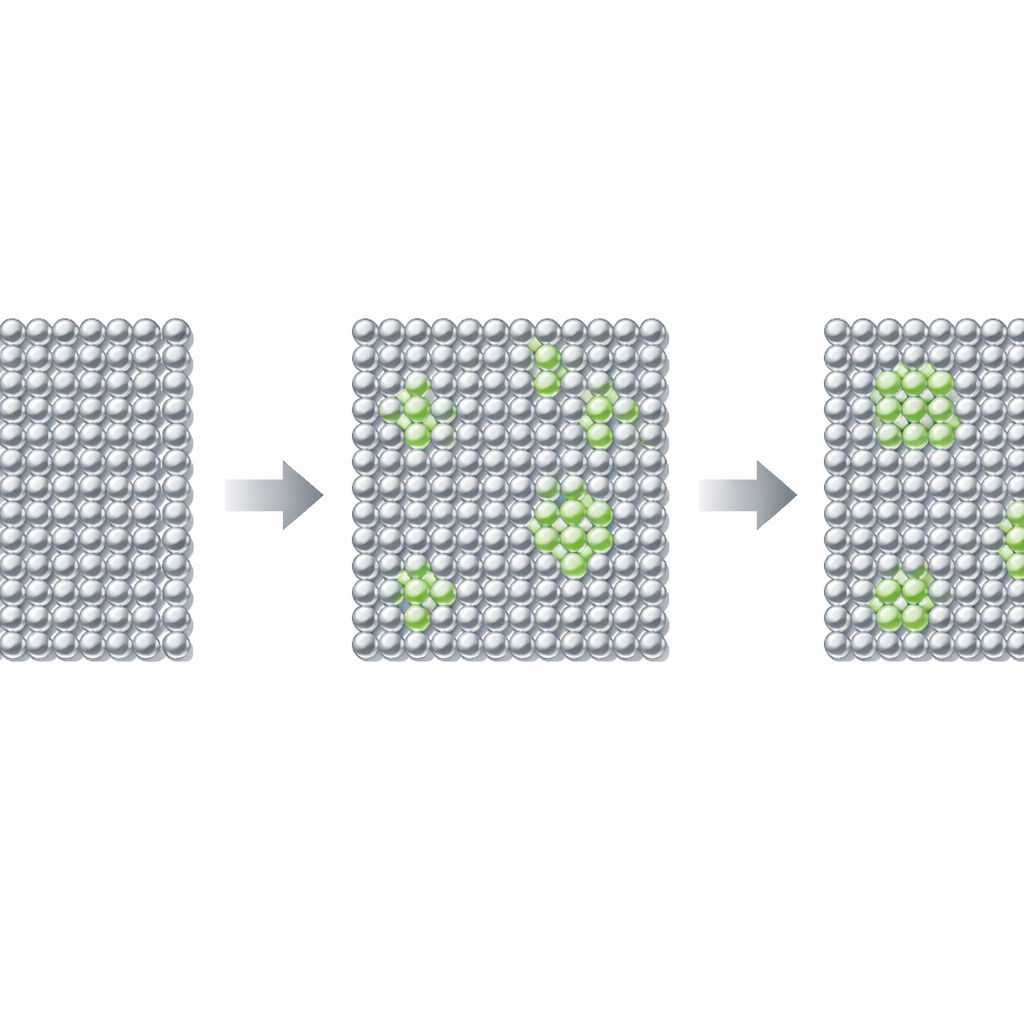
Les étapes cachées à l’intérieur de l’aluminium‑lithium
Après la fabrication, les alliages aluminium‑lithium commencent comme une solution solide uniforme : les atomes de lithium sont dispersés au hasard parmi les atomes d’aluminium. En vieillissant à des températures modérées, les atomes se réarrangent progressivement, traversant plusieurs étapes avant d’atteindre un mélange stable d’aluminium et de particules riches en lithium. Les ingénieurs ont longtemps pensé que des particules sphériques appelées δ′ (de composition proche de Al3Li) apparaissent en premier et assurent une grande partie de la résistance. Mais des expériences ont suggéré une étape encore plus précoce et plus délicate : de très petites régions riches en lithium, baptisées zones GPAl–Li, analogues aux célèbres zones de Guinier–Preston des alliages aluminium‑cuivre classiques. Ces premiers agrégats sont si fugaces et si minuscules que personne n’avait pu établir fermement leur structure ni prouver qu’ils existent réellement en tant que phase distincte.
Simuler les atomes à de multiples échelles
Les auteurs abordent ce problème avec une chaîne de modèles informatiques reliant le comportement quantique aux microstructures visibles au microscope. D’abord, ils utilisent la théorie de la fonctionnelle de la densité, une méthode quantique, pour calculer l’énergie de nombreuses configurations possibles d’atomes d’aluminium et de lithium sur un réseau cubique à faces centrées semblable à celui de l’aluminium pur. Ils entraînent ensuite un modèle d’expansion en clusters, une description mathématique compacte capable d’estimer rapidement l’énergie de nouvelles configurations. Au‑dessus de cela, ils exécutent une méthode de Monte‑Carlo spécialisée, enrichie par de la métadynamique, pour cartographier la façon dont l’énergie libre de l’alliage varie avec la teneur en lithium et la température — construisant essentiellement un « paysage » détaillé qui montre quels motifs atomiques sont favorisés.
Découverte d’un agrégat de lithium ordonné
Ce paysage énergétique révèle un creux distinct autour de 12,5 % atomique de lithium, signalant une configuration métastable : la zone GPAl–Li. En inspectant le motif atomique à cette composition, l’équipe identifie une structure bien ordonnée qu’ils notent δ″ (proche de Al7Li), dans laquelle les atomes de lithium occupent des sites spécifiques du réseau d’aluminium tout en évitant soigneusement d’être voisins directs les uns des autres. L’analyse de la structure électronique explique pourquoi cette disposition est favorisée : le lithium cède des électrons aux atomes d’aluminium voisins d’une manière qui stabilise certaines liaisons, mais uniquement si les atomes de lithium sont espacés de façon appropriée. Les auteurs substituent systématiquement du lithium dans différentes positions voisines et suivent à la fois le nombre d’électrons et les énergies, démontrant que la configuration correspondant à la zone GPAl–Li est un véritable minimum local d’énergie et non un artefact numérique.
Des premiers agrégats aux particules renforçantes
À partir de courbes d’énergie libre précises, les chercheurs construisent ensuite un diagramme de phases métastable incluant la solution solide, les zones GPAl–Li et les précipités δ′ sous la contrainte d’un réseau restant de type aluminium. Ils calculent l’énergie d’interface entre les particules δ′ et la matrice d’aluminium, puis injectent toutes ces informations dans un modèle champ de phase qui simule la diffusion du lithium et l’apparition et la croissance de nouvelles phases en trois dimensions au fil du temps. Ces simulations montrent que, pour une plage utile de teneurs en lithium et pour des températures inférieures à environ 483 K (environ 210 °C), l’alliage forme d’abord des zones GPAl–Li étendues, qui se transforment ensuite en particules δ′. Près de la composition idéale de la zone GPAl–Li, la présence d’un puits d’énergie local profond ralentit en fait la croissance des δ′, expliquant des rapports expérimentaux où un contenu en lithium plus élevé ne conduisait pas systématiquement à un durcissement plus rapide.
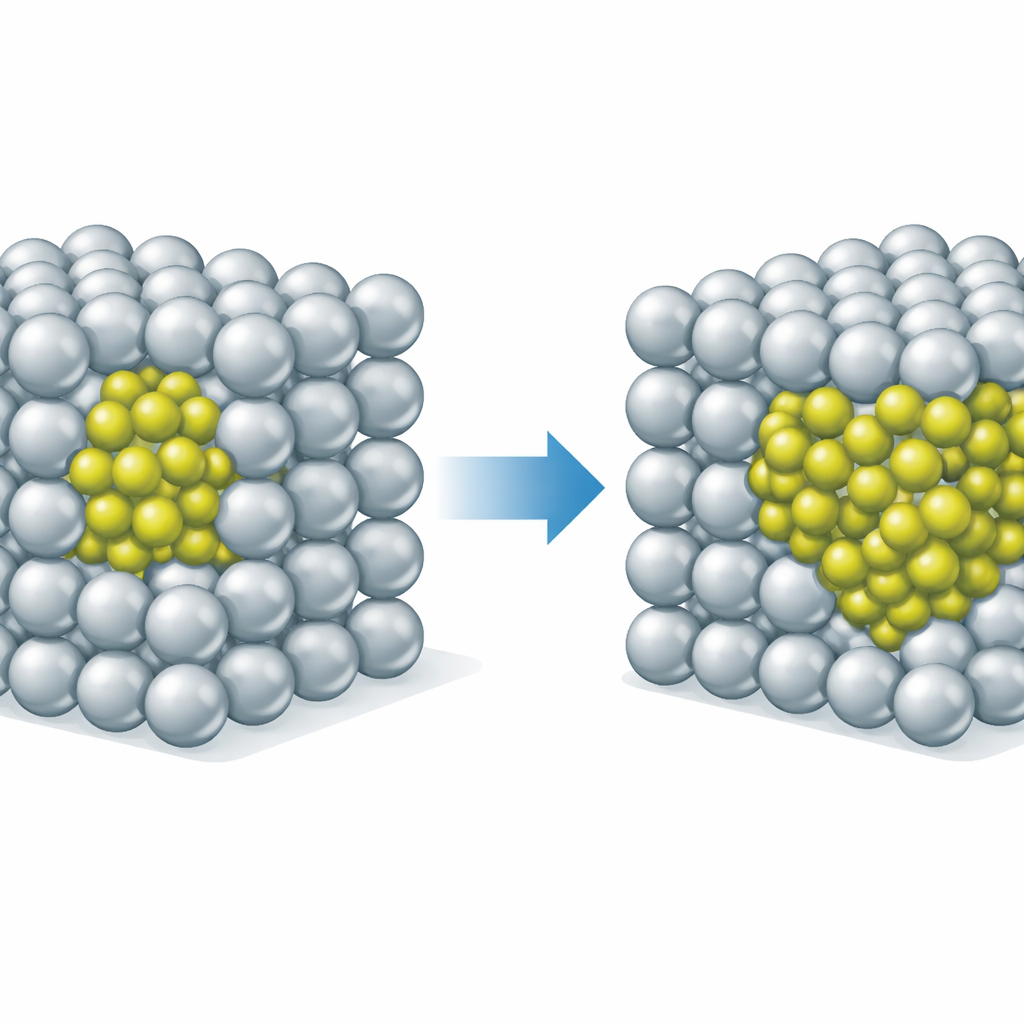
Pourquoi les traitements cryogéniques et les ajouts de cuivre importent
La modélisation clarifie aussi pourquoi les zones GPAl–Li sont si difficiles à surprendre in situ. À température ambiante et au‑dessus, ces zones sont seulement brièvement métastables et évoluent rapidement en δ′, laissant peu de preuves directes. À des températures cryogéniques, cependant, la diffusion du lithium est beaucoup plus lente tandis que le puits d’énergie pour la structure GPAl–Li s’approfondit, si bien que les zones peuvent persister suffisamment longtemps pour être observées dans des échantillons traités avec soin. Enfin, en considérant comment ces régions riches en lithium interagissent avec le cuivre dans des alliages aluminium‑lithium‑cuivre plus complexes, les auteurs proposent que les zones GPAl–Li peuvent servir de lieux privilégiés pour la formation des plaques de renforcement importantes T1 (Al2CuLi). Cette perspective suggère de nouvelles stratégies de composition et de traitements thermiques pour concevoir des alliages aéronautiques plus légers et plus résistants.
Ce que cela signifie pour les alliages réels
En résumé, l’étude montre que la mystérieuse zone GPAl–Li est une configuration atomique ordonnée réelle qui apparaît brièvement entre l’alliage initialement uniforme et les particules δ′ connues. En cartographiant quand et comment cette étape se forme et se transforme, ce travail comble une lacune cruciale dans l’histoire du durcissement des alliages aluminium‑lithium. Pour les ingénieurs, cela se traduit par des recettes plus fiables pour la composition des alliages et les traitements thermiques — en particulier à basse température et pour des alliages contenant aussi du cuivre — ouvrant la voie à des structures d’avions et de fusées plus légères et plus sûres.
Citation: Tian, Q., Hou, L., Wang, J. et al. Multi-scale modeling GPAl-Li zones in Al-Li alloys starting from first-principles. npj Comput Mater 12, 104 (2026). https://doi.org/10.1038/s41524-026-01974-6
Mots-clés: alliages aluminium‑lithium, durcissement par précipitation, zones de Guinier‑Preston, matériaux assistés par calcul, simulation champ de phase