Clear Sky Science · fr
Flux de travail automatisé assisté par un potentiel d’apprentissage automatique auto‑optimisant pour la conception de matériaux de systèmes complexes très efficaces
Des recherches plus intelligentes pour de nouveaux matériaux
Concevoir de nouveaux matériaux revient un peu à chercher une aiguille dans une meule de foin quasi infinie. Des batteries plus performantes et des ordinateurs plus rapides aux lasers plus efficaces et aux éventuels supraconducteurs à température ambiante, de nombreuses technologies futures dépendent de la découverte d’arrangements atomiques précis. Cet article propose une méthode pour confier à l’intelligence artificielle la majorité de cette recherche de façon automatique, réduisant considérablement le temps et le coût nécessaires pour trouver des composés prometteurs.
Pourquoi le casse‑tête des matériaux est si difficile
Les propriétés d’un solide — sa conductivité électrique, sa résistance, sa réponse à la lumière — sont déterminées par la façon dont ses atomes sont disposés dans des motifs tridimensionnels appelés structures cristallines. En théorie, on peut utiliser la mécanique quantique pour calculer quelles configurations sont stables et quelles seront leurs propriétés. En pratique, ces calculs quantiques sont si coûteux qu’une infime fraction seulement des matériaux possibles peut être examinée. Le défi augmente rapidement lorsqu’on considère plus de deux éléments chimiques, car le nombre de combinaisons et d’agencements atomiques explose, rendant une recherche aveugle impraticable.
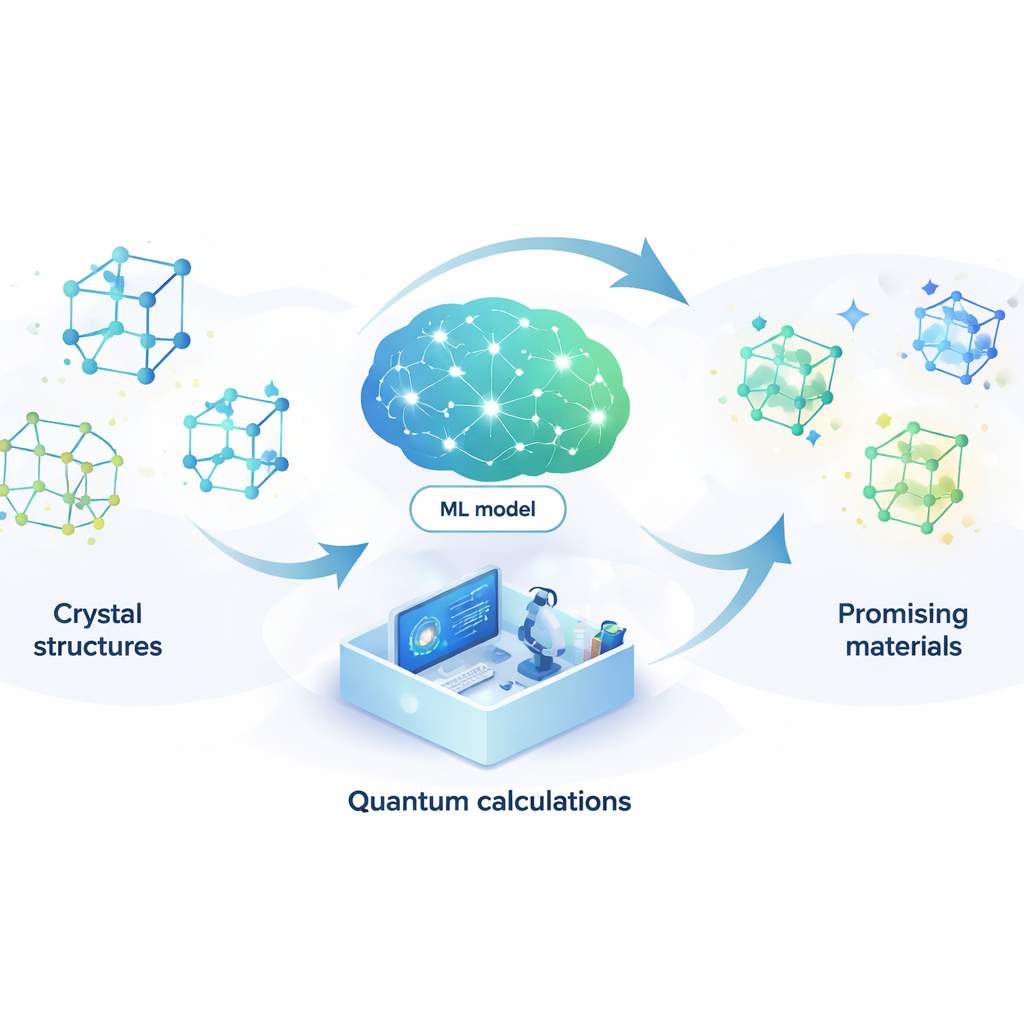
Laisser un modèle d’apprentissage remplacer la physique quantique
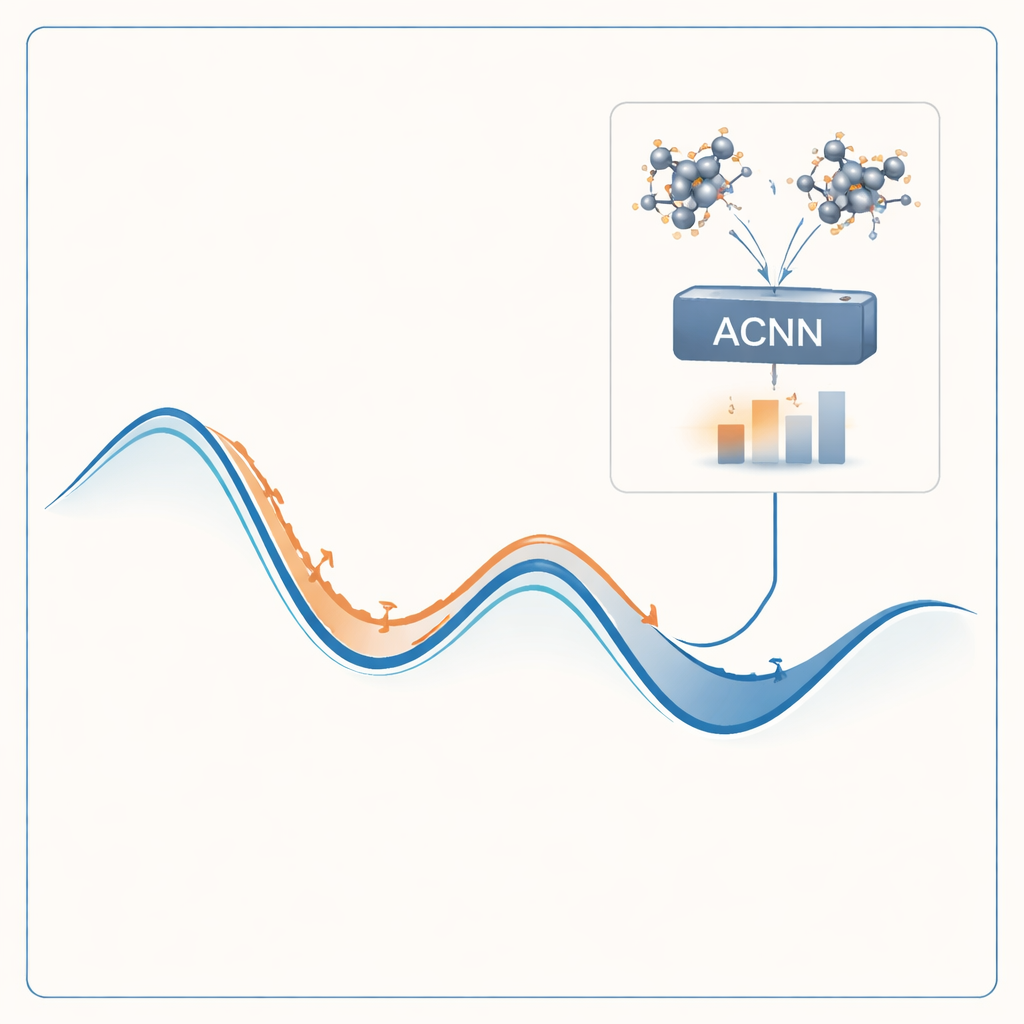
Pour s’attaquer à ce problème, les auteurs construisent un modèle d’apprentissage automatique capable d’imiter les résultats des coûteux calculs quantiques à une fraction infime du coût. Leur modèle, appelé réseau neuronal couplé par attention (ACNN), apprend comment l’énergie d’un matériau dépend des positions et des types d’atomes. Une fois entraîné, il peut estimer très rapidement si une structure cristalline proposée est probablement stable ou non, et quelles forces s’exercent sur chaque atome. Surtout, le modèle est conçu pour respecter des contraintes physiques fondamentales, comme le fait que le déplacement ou la rotation de l’ensemble du cristal ne doit pas modifier son énergie totale.
Une boucle d’amélioration automatique pour la découverte de matériaux
Plutôt que d’entraîner le modèle une fois pour toutes en espérant qu’il fonctionne partout, les auteurs l’intègrent dans une boucle auto‑optimisante. Le processus commence par un petit ensemble de structures cristallines aléatoires, évaluées par des calculs quantiques complets et utilisées pour entraîner un ACNN initial. Ce modèle sert ensuite à relaxer des millions de structures candidates, trouvant rapidement des minima d’énergie locaux — des phases potentiellement stables ou quasi‑stables. Le flux de travail repère automatiquement deux types de structures particulièrement utiles : celles qui paraissent très stables et celles qui semblent non physiques ou suspectes. Seuls ces cas sélectionnés sont renvoyés au solveur quantique coûteux, et les nouveaux résultats sont réintégrés pour réentraîner le modèle. Au fil de nombreuses itérations, le modèle devient de plus en plus précis dans les régions de l’espace des structures qui comptent le plus.
Mettre la méthode à l’épreuve
L’équipe illustre son approche sur deux systèmes exigeants. Le premier est un mélange à haute pression de magnésium, calcium et hydrogène, une famille de composés d’un grand intérêt pour la supraconductivité à haute température. En explorant près de six millions de structures candidates, leur flux de travail dévoile une nouvelle phase stable, MgCa₃H₂₃, et plusieurs structures « en cage » riches en hydrogène apparentées. Les calculs suggèrent que certaines d’entre elles pourraient être supraconductrices à des températures supérieures au point d’ébullition de l’azote liquide sous des pressions extrêmes. Le deuxième test porte sur un système à quatre éléments contenant béryllium, phosphore, azote et oxygène, choisi pour son potentiel à héberger des cristaux capables de convertir efficacement la lumière laser en longueurs d’onde ultraviolettes profondes. Là, la méthode a relaxé plus de neuf millions de structures et identifié trois phases thermodynamiquement stables aux larges gaps de bande et aux caractéristiques optiques prometteuses.
De la force brute à la découverte guidée
Dans les deux exemples, le flux de travail automatisé atteint des accélérations d’environ dix mille fois par rapport à l’utilisation exclusive de calculs quantiques, tout en identifiant de manière fiable les structures dignes d’un examen approfondi. Pour le non‑spécialiste, le message clé est qu’une grande partie de la découverte de matériaux peut désormais être prise en charge par un système d’apprentissage qui apprend lui‑même où il est incertain et sollicite des calculs ciblés et de haute précision uniquement quand cela est nécessaire. Ce type de recherche auto‑corrigée et assistée par IA ouvre la voie à l’exploration de mélanges d’éléments bien plus complexes qu’auparavant, augmentant les chances de découvrir de nouveaux supraconducteurs, cristaux optiques et autres matériaux fonctionnels qui soutiendront les technologies de prochaine génération.
Citation: Li, J., Feng, J., Luo, J. et al. Self-optimizing machine learning potential assisted automated workflow for highly efficient complex systems material design. npj Comput Mater 12, 101 (2026). https://doi.org/10.1038/s41524-026-01971-9
Mots-clés: découverte de matériaux, potentiels d’apprentissage automatique, prédiction de structures cristallines, hydrides supraconducteurs, cristaux optiques non linéaires