Clear Sky Science · fr
Apprendre une dynamique moléculaire grossière efficace en données à partir des forces et du bruit
Pourquoi réduire les molécules importe
Simuler le mouvement incessant de chaque atome d’une protéine et de l’eau qui l’entoure est l’un de nos meilleurs outils pour comprendre comment la vie fonctionne à l’échelle moléculaire. Mais ces simulations tout‑atomes sont si gourmandes en calcul qu’il peut falloir des mois sur un superordinateur pour suivre une protéine qui se replie, se déplie ou interagit avec des partenaires sur des échelles de temps biologiquement pertinentes. Cet article présente une nouvelle façon de construire des modèles simplifiés et rapides de protéines qui conservent un comportement proche de leurs équivalents atomiques, tout en nécessitant beaucoup moins de données d’entraînement et de puissance de calcul qu’auparavant.
De chaque atome à une image simplifiée
La dynamique moléculaire traditionnelle suit chaque atome et calcule les forces entre eux à chaque pas de temps infinitésimal. Pour accélérer les calculs, les chercheurs utilisent souvent des modèles grossiers (coarse‑grained) qui regroupent de nombreux atomes en un nombre réduit de « billes ». Ces modèles réduits s’exécutent beaucoup plus vite mais ont historiquement eu du mal à égaler la précision des simulations atomistiques complètes, en particulier pour des protéines au repliement riche. Des travaux récents ont recours au machine learning pour découvrir automatiquement de meilleurs champs de forces grossiers, mais l’apprentissage de ces modèles a généralement exigé des millions d’instantanés détaillés, chacun annoté par les forces sur chaque atome — une charge énorme en données et en calcul.
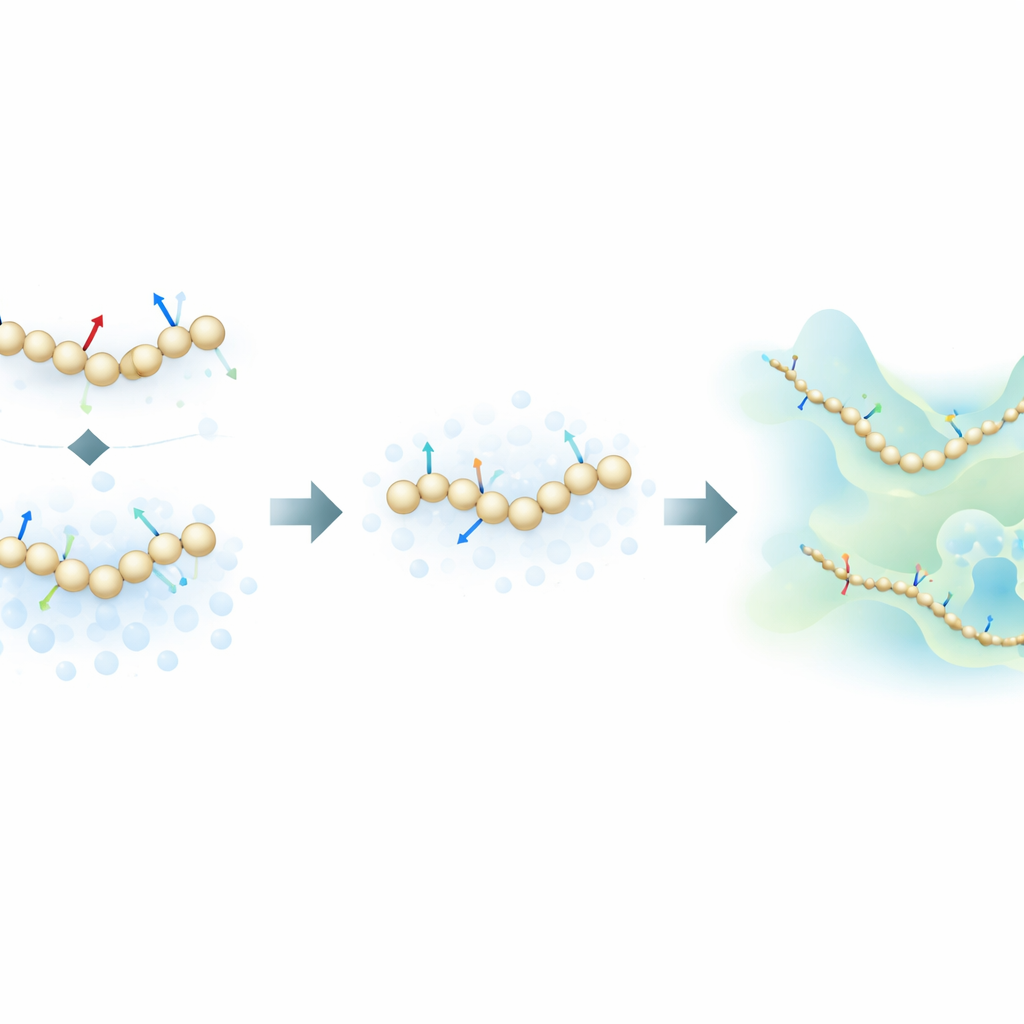
Mêler forces physiques et bruit informatif
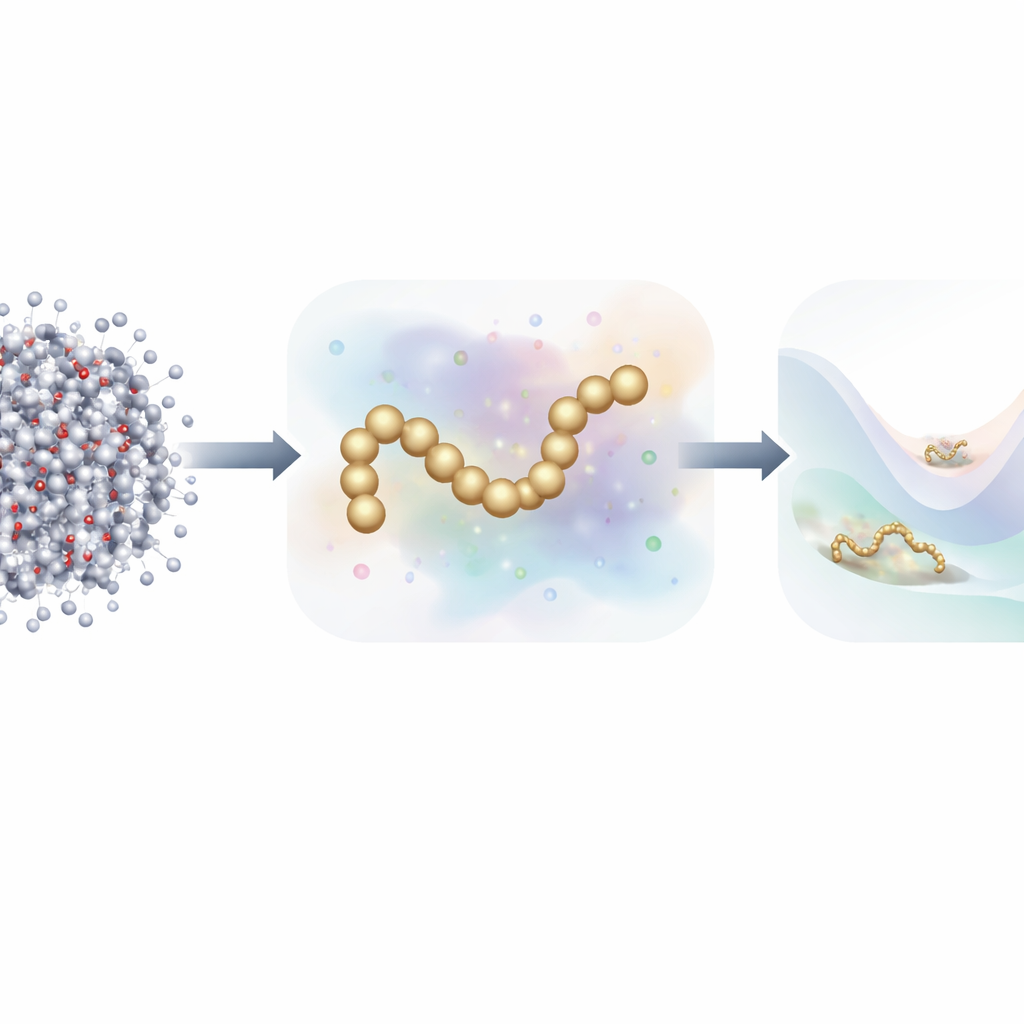
Les auteurs proposent une nouvelle stratégie d’entraînement qui s’inspire des modèles génératifs par diffusion — la même classe d’algorithmes derrière de nombreux générateurs d’images IA modernes. Plutôt que d’apprendre uniquement à partir des forces physiques calculées dans les simulations atomistiques, leur méthode apprend aussi à partir de la distribution des structures moléculaires en ajoutant délibérément du bruit contrôlé aux configurations coarse‑grained. Dans ce cadre, le bruit n’est pas seulement une nuisance à éliminer ; il devient une source d’information supplémentaire. En unifiant mathématiquement l’approche traditionnelle de « mise en correspondance des forces » (force matching) avec des techniques de débruitage issues des modèles de diffusion, la méthode peut inférer le paysage énergétique sous‑jacent d’une protéine en utilisant beaucoup moins d’exemples annotés.
Apprendre à des modèles simples à imiter des protéines complexes
Pour tester leur idée, les chercheurs ont entraîné des modèles coarse‑grained à réseau de neurones pour plusieurs protéines d’une complexité croissante : les petites miniprotéines Chignolin et Trp‑Cage, la NTL9 un peu plus grande, et l’ubiquitine de 76 résidus. Ils ont comparé trois modes d’entraînement : n’utiliser que les forces atomistiques, n’utiliser que l’information issue du bruit, et combiner les deux. Pour les protéines les plus petites, ils montrent que la nouvelle approche combinée peut reproduire les caractéristiques clés du paysage de repliement — comme la stabilité relative des états repliés et déroulés et la présence d’intermédiaires — en nécessitant jusqu’à cent fois moins de données d’entraînement que les méthodes traditionnelles de force matching. De manière surprenante, dans les régimes pauvres en données, même les modèles entraînés uniquement avec l’information issue du bruit égalent ou dépassent souvent la précision de l’entraînement basé uniquement sur les forces.
Atteindre des systèmes protéiques plus grands et plus difficiles
L’ubiquitine constitue un test plus exigeant : capturer son repliement et son déroulement à des températures réalistes a historiquement nécessité du matériel spécialisé et des simulations atomistiques extrêmement longues. Ici, les auteurs entraînent des modèles coarse‑grained en utilisant un jeu de données modeste composé de courtes simulations d’équilibre autour de l’état replié ainsi que de simulations non‑équilibre « tirées » qui étirent la protéine de force. Malgré cet ensemble d’entraînement biaisé et l’absence d’une référence atomistique parfaite aux mêmes conditions, le modèle entraîné avec forces et bruit retrouve une image réaliste où états repliés et déroulés coexistent, l’état replié étant favorisé en stabilité. En revanche, un modèle entraîné uniquement sur les forces échoue à stabiliser l’état replié, tandis qu’un modèle uniquement basé sur le bruit préfère des structures déroulées. Notamment, aucun des modèles coarse‑grained n’a simplement mémorisé les formes étirées extrêmes issues des données d’entraînement, ce qui indique que le paysage énergétique appris a un sens physique et n’est pas seulement l’empreinte des trajectoires d’entrée.
Ce que cela signifie pour les simulations à venir
En transformant le bruit en signal d’entraînement et en le fusionnant avec les forces physiques, ce travail montre qu’il est possible de construire des modèles coarse‑grained précis pour les protéines à partir de jeux de données bien plus petits et moins parfaits qu’on ne le pensait. En pratique, cela signifie que les chercheurs n’auront peut‑être plus besoin de simulations atomistiques de l’ordre de la milliseconde sur des superordinateurs spécialisés avant d’explorer le comportement d’une biomolécule avec une dynamique coarse‑grained apprise par machine. À la place, des simulations plus modestes sur du matériel largement disponible pourraient suffire à entraîner des modèles réduits puissants qui capturent les principales voies de repliement et les équilibres thermodynamiques. Si des questions subsistent sur le meilleur choix et l’interprétation du bruit ajouté et sur la performance de la méthode pour des assemblages biomoléculaires encore plus grands et complexes, cette approche abaisse sensiblement la barrière à l’usage routinier de simulations coarse‑grained pilotées par les données en science moléculaire.
Citation: Durumeric, A.E.P., Chen, Y., Pasos-Trejo, A.S. et al. Learning data-efficient coarse-grained molecular dynamics from forces and noise. Nat Commun 17, 2493 (2026). https://doi.org/10.1038/s41467-026-70818-0
Mots-clés: dynamique moléculaire grossière, champs de forces appris par machine learning, simulations du repliement des protéines, modèles de diffusion en chimie, simulation économe en données