Clear Sky Science · fr
Restaurer la dysrégulation synaptique postnatale précoce sauve la dégénérescence des motoneurones dans un modèle murin de l’atrophie musculaire spinale et bulbaire
Pourquoi de minuscules changements précoces peuvent avoir des conséquences sur une faiblesse musculaire ultérieure
L’atrophie musculaire spinale et bulbaire (SBMA) est une maladie héréditaire rare dans laquelle des adultes, généralement des hommes, perdent progressivement de la force dans les membres, le tronc et la gorge. Les symptômes apparaissent à l’âge adulte, mais des troubles subtils commencent bien plus tôt. Cette étude pose une question surprenante : de brefs événements survenant dans les premiers jours après la naissance pourraient-ils discrètement préparer le terrain pour la perte de cellules nerveuses des décennies plus tard — et corriger ces anomalies précoces pourrait-il protéger le mouvement ?
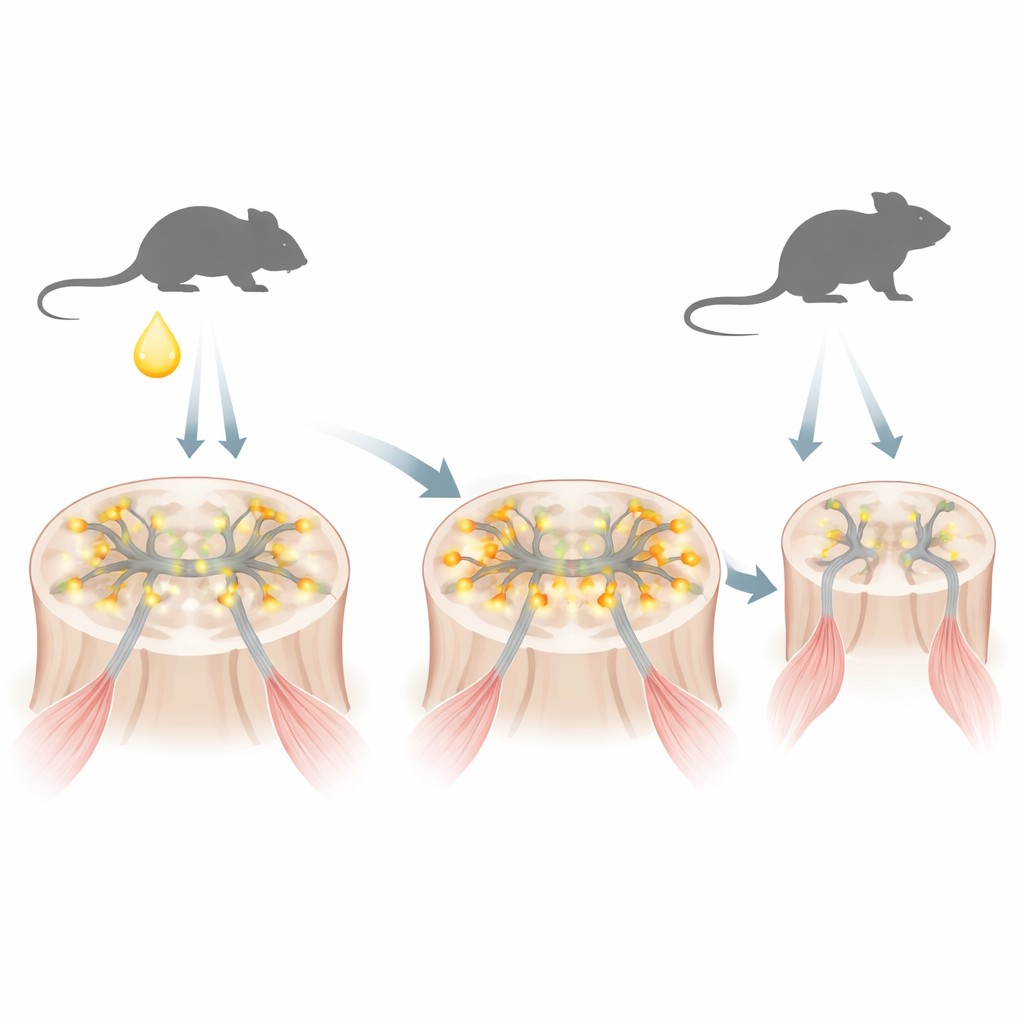
Une maladie ancrée dans un interrupteur sensible aux hormones
La SBMA est causée par une version altérée du récepteur aux androgènes, une protéine qui détecte les hormones mâles comme la testostérone. Le récepteur muté porte une longue répétition excessive de l’acide aminé glutamine. Dans un modèle murin qui reproduit la maladie humaine, les auteurs ont observé qu’immédiatement après la naissance, lorsque la testostérone augmente brièvement chez les mâles nouveau‑nés, ce récepteur mutant s’accumule rapidement dans les noyaux des motoneurones — les cellules nerveuses qui commandent les muscles. À ce stade précoce, la protéine ne s’est pas encore agrégée en amas volumineux typiquement associés à la neurodégénérescence, mais elle modifie déjà l’expression de certains gènes.
Surtension synaptique précoce et motoneurones agités
En utilisant le séquençage ARN à l’échelle du génome sur les moelles épinières de souris nouveau‑nées, l’équipe a découvert que de nombreux gènes impliqués dans les synapses excitatrices — points de contact où les neurones communiquent — étaient anormalement actifs. Beaucoup de ces gènes codent pour des récepteurs au glutamate, qui rendent les neurones plus susceptibles de déclencher des potentiels d’action. Le groupe a lié ce profil à une altération de REST, une protéine « frein » maîtresse qui, en développement, maintient normalement ces gènes synaptiques sous contrôle strict. Chez les souris SBMA et dans des motoneurones dérivés de cellules souches pluripotentes induites de patients, l’activité de REST était réduite et une forme raccourcie appelée REST4 était favorisée, levant le frein et augmentant l’expression des gènes des synapses glutamatergiques. Conforme à cela, les motoneurones SBMA nouveau‑nés présentaient des niveaux plus élevés de c‑Fos, un marqueur d’activité récente, et les motoneurones humains dérivés de patients montraient des bouffées calciques plus fortes et plus fréquentes, signes d’hyperexcitabilité.
Un traitement bref en période néonatale modifiant le cours à vie
Les chercheurs ont ensuite demandé si réduire le récepteur mutant ou restaurer le frein REST uniquement pendant cette fenêtre néonatale pouvait modifier l’évolution à long terme de la maladie. Ils ont administré des oligonucléotides antisens — de courts brins d’acide nucléique modifié — dans le liquide entourant le cerveau et la moelle épinière de souris SBMA d’un jour. Un type d’oligonucléotide abaissait temporairement à la fois le récepteur aux androgènes mutant et la forme normale dans le système nerveux central. Un second type orientait l’épissage de REST loin de REST4 et vers la forme complète de REST, réprimant ainsi les gènes synaptiques. De manière remarquable, bien que ces traitements n’aient été administrés qu’une seule fois et que leurs effets moléculaires directs se soient estompés en quelques semaines, les souris ont vécu plus longtemps, mieux marché sur une barre rotative et gardé une préhension plus forte à l’âge adulte. Leurs motoneurones et fibres musculaires étaient moins atrophiés, et les marqueurs précoces de suractivité neuronale ainsi que les pics ultérieurs de neuropeptides liés au stress étaient atténués.
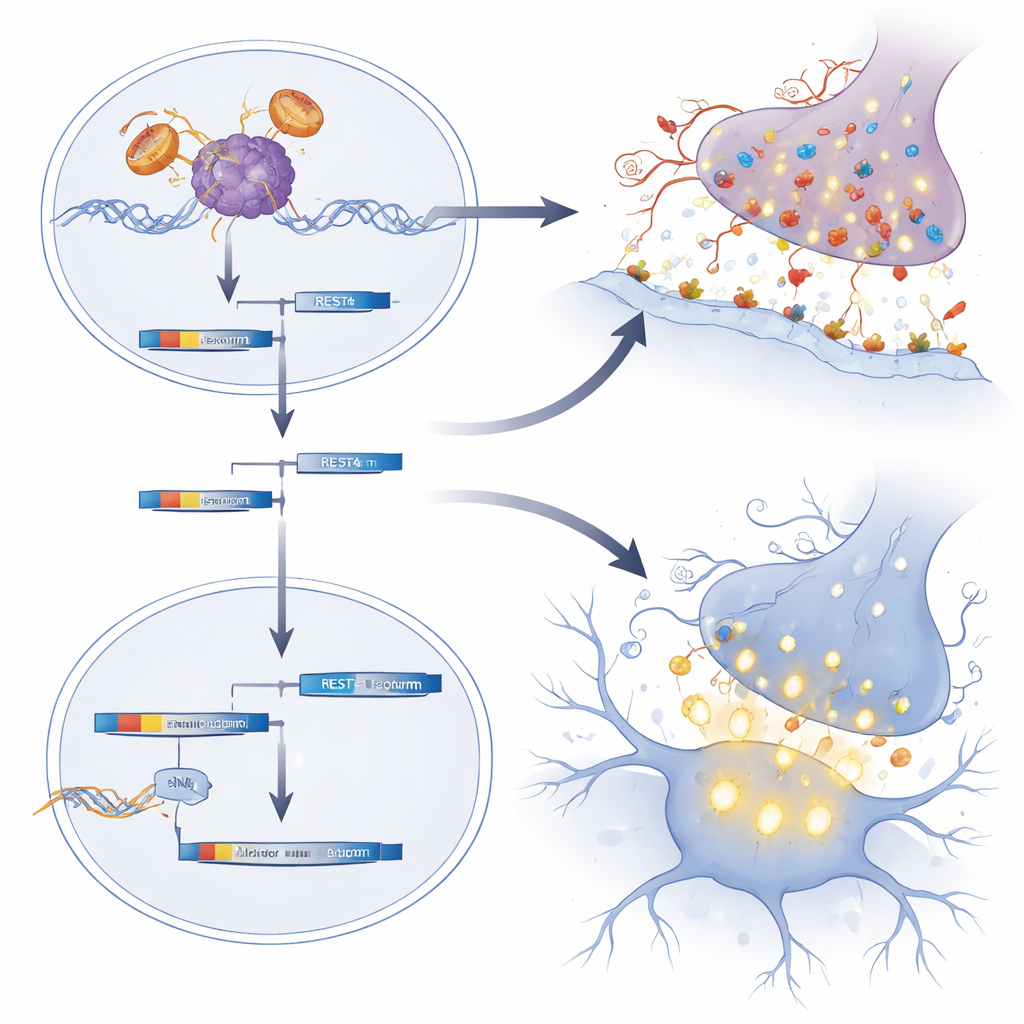
Comment les hormones précoces et le réglage génétique conditionnent la vulnérabilité
Ce travail met aussi en évidence la vulnérabilité particulière des motoneurones à la brève poussée de testostérone qui survient peu après la naissance. Lorsque des souris SBMA nouveau‑nées ont reçu un supplément de testostérone, leur faiblesse ultérieure et la perte de poids se sont aggravées, et les programmes géniques liés à une maturation saine des motoneurones ont été encore plus perturbés. Les souris normales n’ont pas montré ces lésions, ce qui souligne que c’est la combinaison du récepteur mutant et de la poussée hormonale qui est délétère. Ensemble, ces résultats suggèrent que dans la SBMA, un excès de synapses excitatrices et des motoneurones trop excitable précocement poussent lentement le système vers l’échec, même si des symptômes évidents n’apparaissent qu’à l’âge moyen.
Ce que cela signifie pour les personnes vivant avec la SBMA
Pour un non‑spécialiste, le message clé est que la SBMA peut être, en partie, une maladie de synapses mal synchronisées et mal câblées pendant les premiers jours après la naissance. Un capteur hormonal défectueux pousse les motoneurones en développement dans un état de surexcitation, et ce stress précoce contribue finalement à leur dégénérescence des années plus tard. La nouvelle encourageante est que des médicaments génétiques soigneusement conçus, administrés pendant ces fenêtres critiques, peuvent rééquilibrer les signaux dans les motoneurones, calmer leur suractivité et retarder ou réduire de manière significative la perte ultérieure de cellules nerveuses chez l’animal. La transposition de telles interventions en vie humaine exigera beaucoup de prudence et des études supplémentaires, mais ces résultats ouvrent la voie à de nouvelles stratégies visant les racines de la SBMA bien avant l’apparition de la faiblesse.
Citation: Hirunagi, T., Sahashi, K., Iida, M. et al. Restoring early postnatal synaptic dysregulation rescues motor neuron degeneration in a mouse model of Spinal and Bulbar Muscular Atrophy. Nat Commun 17, 2412 (2026). https://doi.org/10.1038/s41467-026-70244-2
Mots-clés: atrophie musculaire spinale et bulbaire, hyperexcitabilité des motoneurones, récepteur aux androgènes, régulation synaptique REST, thérapie par oligonucléotides antisens