Clear Sky Science · fr
DiNovo permet un séquençage de novo des peptides à haute couverture et haute confiance grâce à des protéases miroir et à l’apprentissage profond
Voir les protéines avec plus de précision
Les protéines sont les petites machines qui maintiennent nos cellules en vie, mais lire complètement leurs éléments constitutifs reste étonnamment difficile. Cet article présente DiNovo, un nouveau système logiciel qui aide les scientifiques à « lire » les fragments protéiques de manière beaucoup plus complète et fiable qu’auparavant. En combinant une astuce biochimique ingénieuse avec l’intelligence artificielle moderne, il promet de révéler des protéines cachées, des marqueurs de maladie et même des cibles immunitaires que les méthodes traditionnelles manquent souvent.
Pourquoi lire les fragments protéiques est si difficile
La plupart des analyses protéiques actuelles reposent sur la découpe des protéines en morceaux plus petits, appelés peptides, puis sur la mesure de leurs fragments dans un spectromètre de masse. À partir de ces masses, des ordinateurs tentent de reconstruire la séquence peptidique originale, comme résoudre un mot croisé à partir d’indices partiels. Les méthodes existantes supposent généralement que les peptides proviennent de bases de données protéiques connues, ce qui fonctionne bien pour des protéines familières mais peine face à des protéines nouvelles ou inattendues. Le séquençage dit de novo évite cette limitation en essayant de lire les peptides directement à partir des données, mais il échoue souvent parce que certains fragments manquent et que certains peptides ne sont jamais coupés proprement dès le départ.
Utiliser des enzymes miroir pour combler les lacunes
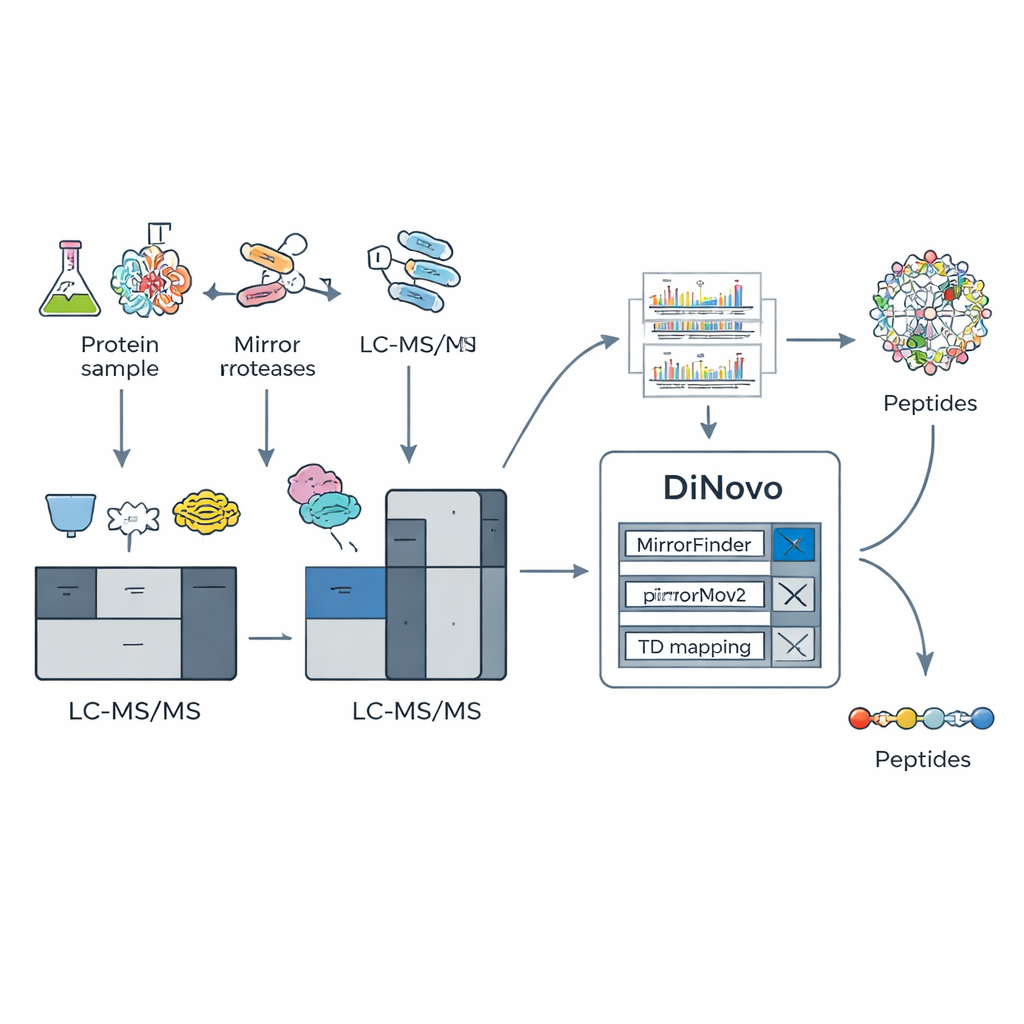
L’idée clé derrière DiNovo est d’utiliser des paires de « protéases miroir » – des paires d’enzymes de coupure qui clivent les protéines de part et d’autre du même type d’acide aminé. Par exemple, une enzyme coupe juste avant une lysine, tandis que sa partenaire coupe juste après cette lysine. Cela produit deux peptides apparentés qui partagent le même segment interne mais ont des extrémités différentes. Lorsque ces peptides « miroir » sont analysés, leurs spectres de masse contiennent des motifs de fragments complémentaires : ce qui manque dans un spectre apparaît souvent dans l’autre. Les auteurs montrent que la combinaison de telles paires miroir peut porter la couverture des fragments proche de l’exhaustivité, avec environ 98 % des coupures possibles appuyées par des signaux expérimentaux réels, bien plus que ce que l’on obtient avec une seule enzyme.
Un pipeline logiciel intelligent conçu pour les données miroir
Pour exploiter cette astuce biochimique, l’équipe a construit DiNovo comme un flux de travail logiciel de bout en bout. D’abord, des protéines de bactéries et de levure sont digérées avec deux paires miroir d’enzymes, et les peptides résultants sont analysés par spectrométrie de masse haute résolution. DiNovo utilise ensuite un module appelé MirrorFinder pour reconnaître automatiquement quelles paires de spectres proviennent de peptides miroir, en se basant directement sur les motifs de signal plutôt que sur des hypothèses de séquence préalables. Ensuite, son moteur principal de novo, MirrorNovo, utilise l’apprentissage profond pour interpréter ces spectres appariés, tandis qu’un moteur de secours basé sur un graphe, pNovoM2, offre une option plus rapide fonctionnant uniquement sur CPU. Ensemble, ces outils traduisent les pics en séquences d’acides aminés et examinent aussi les spectres individuels qui n’ont pas formé de paires évidentes, extrayant le maximum d’informations possible.
Évaluer la confiance sans s’appuyer sur d’anciennes bases de données
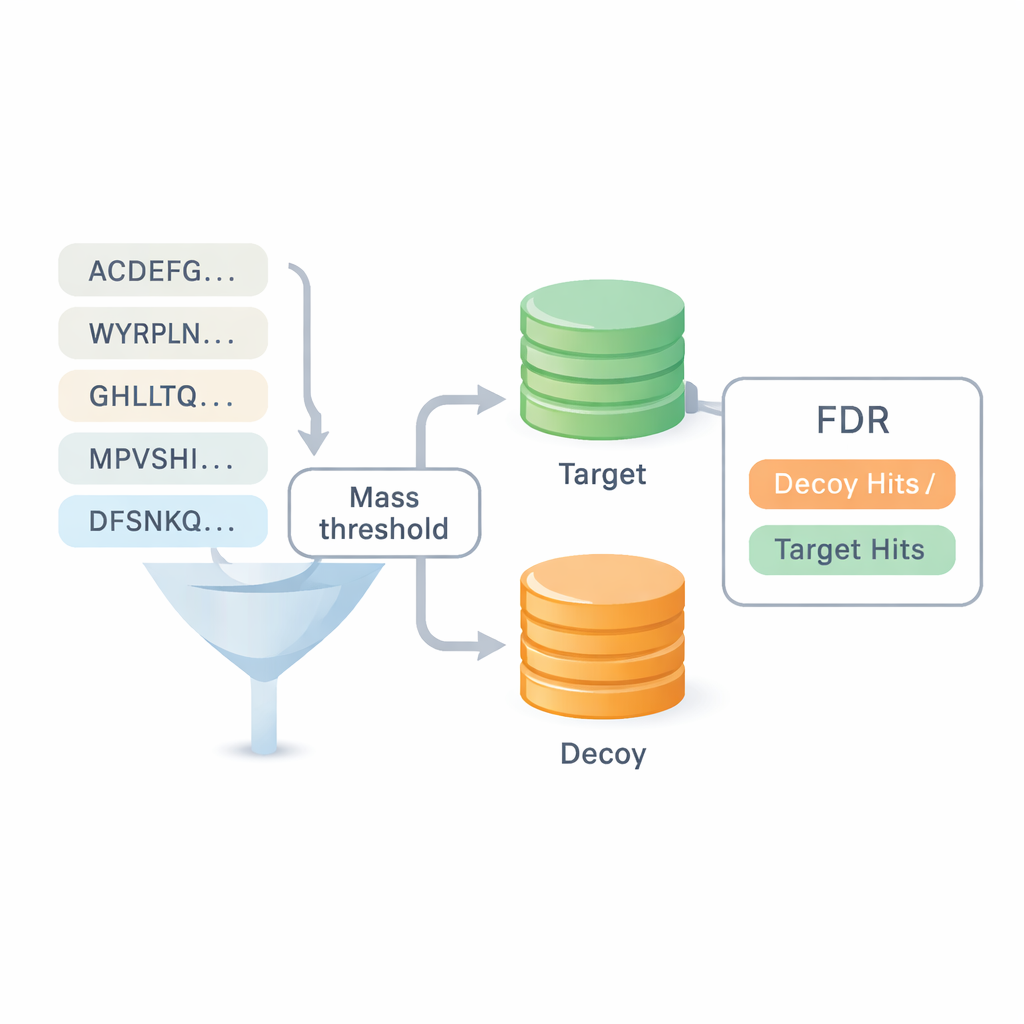
Une des plus grandes questions du séquençage de novo est de savoir combien on peut faire confiance aux résultats. La plupart des benchmarks existants réutilisent les réponses issues de la recherche en base de données, ce qui brouille la frontière entre les deux approches et peut masquer des erreurs. DiNovo introduit une méthode de contrôle qualité différente appelée cartographie cible-leurre (target-decoy mapping). Ici, les peptides nouvellement lus sont cartographiés sur une collection combinée de séquences protéiques réelles (cible) et artificielles, brouillées (leurre). En comparant la fréquence à laquelle les peptides tombent dans l’ensemble réel versus l’ensemble brouillé, le logiciel peut estimer un taux d’erreur, ou taux de fausses découvertes, sans s’appuyer sur des identifications antérieures. Cela rend possible une comparaison directe de DiNovo avec des programmes de recherche en base de données standard sous les mêmes contrôles d’erreur.
Ce que DiNovo apporte en pratique
Dans des tests sur des échantillons bactériens, de levure et d’anticorps, DiNovo a systématiquement lu beaucoup plus de peptides et d’acides aminés que des outils de novo bien connus n’utilisant qu’une seule enzyme. En utilisant deux paires miroir, il a produit 2 à 3 fois plus d’acides aminés à haute confiance qu’un protocole classique basé uniquement sur la trypsine et a identifié davantage de protéines à des niveaux d’erreur similaires. Comparé directement à trois moteurs de recherche en base de données de pointe, DiNovo a trouvé des nombres d’acides aminés et de protéines similaires, et la plupart de ses séquences concordaient avec celles issues des moteurs de recherche sur les mêmes spectres. Les auteurs soutiennent que ce niveau de couverture et d’accord signifie que le séquençage de novo, longtemps considéré comme une méthode de secours, peut désormais se tenir aux côtés de la recherche en base de données comme une option sérieuse et, dans certains cas, supérieure.
Vue d’ensemble : vers une lecture protéique complète et non biaisée
Pour un non-spécialiste, le message est que DiNovo facilite grandement la lecture précise des fragments protéiques sans être limité à ce qui figure déjà dans les bases de référence. En doublant ou triplant la quantité d’informations de séquence bien étayées et en fournissant ses propres contrôles d’erreur intégrés, cette approche ouvre la porte à la découverte de protéines inconnues, au suivi de variations subtiles et à l’exploration de mélanges complexes où de nombreux composants restent inconnus. En bref, en associant des enzymes miroir à l’apprentissage profond et à des statistiques soignées, DiNovo aide à transformer des traces spectraux bruitées en une image plus claire et plus fiable des protéines qui sous-tendent la santé et la maladie.
Citation: Cao, Z., Peng, X., Zhang, D. et al. DiNovo enables high-coverage and high-confidence de novo peptide sequencing via mirror proteases and deep learning. Nat Commun 17, 2203 (2026). https://doi.org/10.1038/s41467-026-70224-6
Mots-clés: protéomique, séquençage de novo des peptides, spectrométrie de masse, apprentissage profond, protéases miroir