Clear Sky Science · fr
La trisomie 21 entraîne une surexpression d'ADARB1 et un re-codage prématuré de l'ARN dans le cerveau fœtal en développement
Comment un chromosome supplémentaire peut reconfigurer le cerveau en développement
Le syndrome de Down est causé par une copie supplémentaire du chromosome 21, mais la manière dont cet excès d'ADN modifie le cerveau en développement est restée mystérieuse. Cette étude examine des cerveaux fœtaux pour voir comment l'activité génique et le « réglage fin » de l'ARN sont altérés avant la naissance. Les travaux se concentrent sur un puissant éditeur des messages ARN, nommé ADARB1, et montrent que son hyperactivité pourrait pousser les cellules cérébrales à maturer prématurément leurs systèmes de communication, ce qui pourrait contribuer à expliquer ultérieurement les différences d'apprentissage et de cognition.
Un regard à l'intérieur du cerveau fœtal
Les chercheurs ont analysé des tissus du cortex préfrontal et de l'hippocampe — deux régions essentielles à la mémoire, à la planification et à l'apprentissage — provenant de fœtus porteurs d'une trisomie 21 et de témoins typiques, tous âgés de 13 à 22 semaines après la conception, une fenêtre clé pour le câblage du cerveau. À l'aide d'un séquençage profond de l'ARN, ils ont mesuré quels gènes étaient activés ou réprimés et comment les molécules d'ARN étaient modifiées chimiquement. Ils ont constaté une perturbation généralisée de l'activité génique en trisomie 21, de nombreux gènes du chromosome 21 étant plus actifs que la normale, comme on pouvait s'y attendre avec une copie supplémentaire. Mais les effets débordaient largement ce seul chromosome, altérant des réseaux impliqués dans l'utilisation d'énergie, la production protéique, ainsi que les fonctions immunitaires et synaptiques. 
Programmes de croissance décalés dans le temps
Un schéma marquant ressemblait à un décalage de développement. Des gènes habituellement très actifs avant la naissance étaient atténués, tandis que des gènes qui s'activent normalement après la naissance s'activaient précocement. Ce décalage apparaissait dans les deux régions cérébrales étudiées et suggérait que les programmes clés guidant la croissance cellulaire, la division et la formation de connexions sont hors de synchronie en trisomie 21. Les groupes de gènes liés à la fonction mitochondriale (les centrales énergétiques de la cellule), à la machinerie de synthèse protéique et au traitement de l'ARN étaient généralement réprimés, alors que ceux associés à la signalisation électrique et à la matrice de soutien du cerveau étaient renforcés. Dans l'hippocampe en particulier, le comportement coordonné habituel des réseaux géniques soutenant la plasticité synaptique, la structure de la chromatine, le métabolisme et les réponses immunitaires était notablement perturbé, suggérant une vulnérabilité spécifique à cette région.
Un éditeur d'ARN hyperactif
Le point central de l'étude est ADARB1, un gène du chromosome 21 qui code pour une enzyme responsable de l'édition adénosine-en-inosine (A-to-I) de l'ARN. Cette modification chimique peut modifier subtilement la séquence et le comportement des protéines ou régler la durée de vie des messages ARN. Dans les cerveaux fœtaux atteints de trisomie 21, les niveaux d'ADARB1 étaient nettement plus élevés, tandis que les enzymes d'édition connexes ne variaient pas. Une mesure globale de l'édition au sein des éléments répétitifs d'ARN était également augmentée, et la modélisation statistique a identifié ADARB1 comme le principal moteur de cette hausse. Lorsque l'équipe a cartographié les sites d'édition individuels à travers le génome, la plupart des changements en trisomie 21 étaient des augmentations d'édition, en particulier dans les régions 3′UTR des ARN, où l'édition peut affaiblir la liaison des microARN régulateurs et déstabiliser les transcrits.
Réglage prématuré des protéines synaptiques
Plus important encore, l'étude a identifié un ensemble de sites classiques de « re-codage » — événements d'édition qui changent la séquence en acides aminés des protéines — au sein de gènes qui construisent des récepteurs au glutamate et au GABA, des régulateurs clés de la signalisation excitatrice et inhibitrice du cerveau. Chez les fœtus avec trisomie 21, des récepteurs codés par des gènes tels que GRIK2, GRIA2, GRIA3 et GABRA3 présentaient des niveaux d'édition supérieurs à la normale en des sites connus pour influencer le flux ionique et la cinétique des récepteurs. En comparant ces niveaux à un large jeu de données de référence du développement cérébral humain typique, les chercheurs ont montré que les fœtus trisomie 21 avaient des profils d'édition ressemblant à ceux observés normalement plus tard dans la vie. Autrement dit, le réglage au niveau de l'ARN de ces récepteurs semblait prématurément avancé. Une méta-analyse de nombreux jeux de données cellulaires et tissulaires indépendants a confirmé une surexpression cohérente d'ADARB1 et une sur-édition à de nombreux sites, en particulier dans les 3′UTR et à un site clé de GRIA3 qui affecte la rapidité de récupération de certains récepteurs après activation. 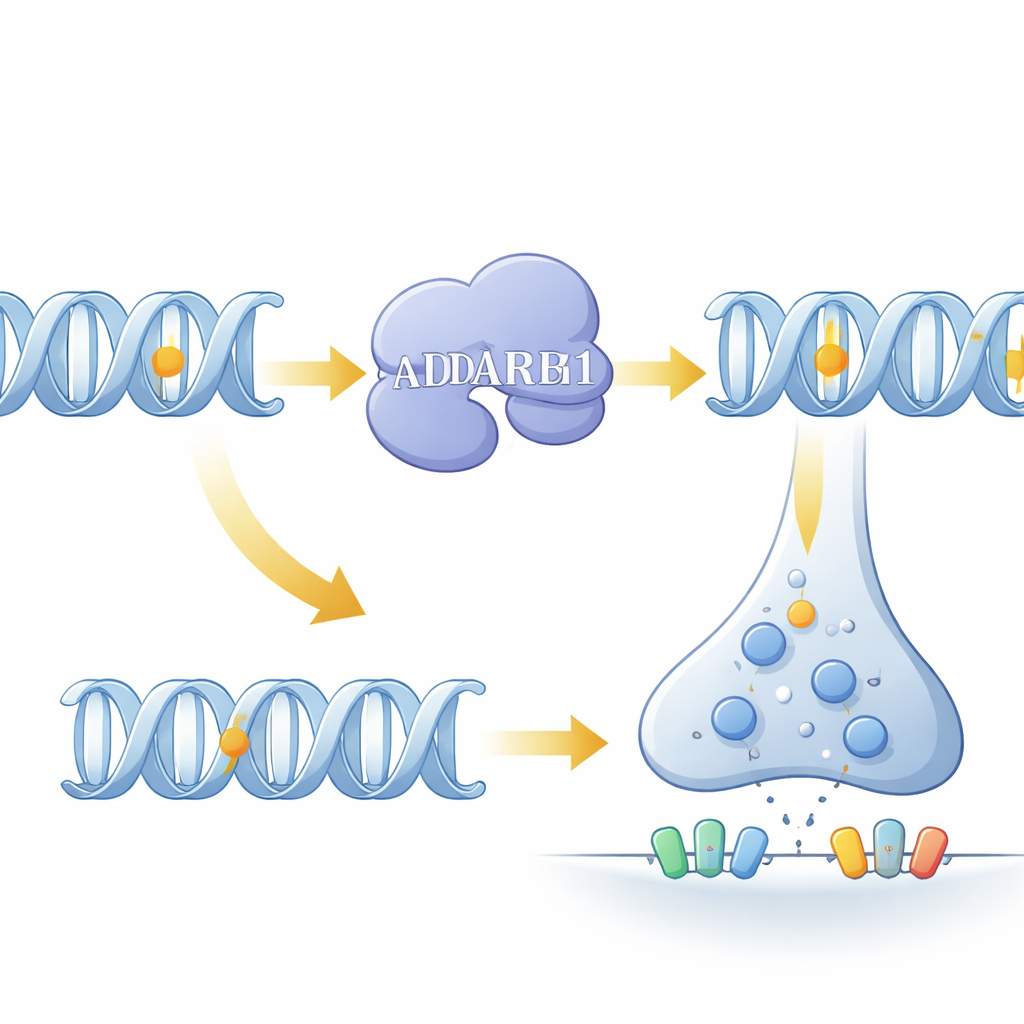
Contexte immunitaire et tissulaire plus large
Parce que la trisomie 21 affecte aussi fortement les voies immunitaires, l'équipe a examiné des échantillons sanguins de centaines d'individus. Là encore, ADARB1 était plus élevé en trisomie 21, mais l'édition globale de l'ARN n'augmentait que chez les personnes dont les cellules immunitaires montraient une forte activation par l'interféron, et cette édition sanguine était surtout due à une enzyme différente, ADAR1. En revanche, dans le cerveau fœtal, les changements d'édition étaient étroitement liés à ADARB1 et à des sites enrichis en neurones, et non à des variations de la composition cellulaire ou des marqueurs immunitaires. Ce contraste souligne que le même chromosome supplémentaire peut remodeler l'édition de l'ARN de façons distinctes dans le cerveau et le système immunitaire.
Que signifie cela pour les personnes atteintes du syndrome de Down
Pour un non-spécialiste, le message principal est qu'une copie supplémentaire du chromosome 21 ne se contente pas de moduler l'expression de certains gènes : elle semble aussi sur-activier un « correcteur » moléculaire de l'ARN, ADARB1, dans le cerveau fœtal. Cette sur-édition accélère le réglage normal des protéines réceptrices qui contrôlent la communication entre cellules cérébrales, ce qui peut amener les circuits à mûrir selon une chronologie décalée et modifier l'équilibre excitation/inhibition. Bien que l'étude n'établisse pas une relation de cause à effet définitive, elle met en évidence l'édition de l'ARN comme une couche puissante et auparavant sous-estimée de la biologie du syndrome de Down — une couche qui pourrait à terme servir à suivre les changements précoces du cerveau ou à guider des thérapies visant à restaurer un calendrier et une intensité de communication neuronale plus typiques.
Citation: Breen, M.S., Yang, A., Wang, X. et al. Trisomy 21 Drives ADARB1 Overexpression and Premature RNA Recoding in the Developing Fetal Brain. Nat Commun 17, 2797 (2026). https://doi.org/10.1038/s41467-026-70217-5
Mots-clés: Syndrome de Down, développement du cerveau fœtal, édition de l'ARN, ADARB1, signalisation synaptique