Clear Sky Science · fr
Échantillonnage efficace des voies de transition à grande échelle et des conformations intermédiaires dans des complexes protéiques sub-mésoscopiques
Observer les protéines en mouvement
Beaucoup des molécules qui nous maintiennent en vie se comportent moins comme des briques Lego rigides que comme de minuscules machines qui changent constamment de forme. Ces mouvements alimentent des processus tels que la production d’énergie, la réparation de l’ADN et l’entrée des virus dans les cellules. Des expériences comme la cryo‑microscopie électronique peuvent désormais figer certaines de ces conformations, mais pas les étapes fugaces intermédiaires. Cet article présente eBDIMS2, une nouvelle méthode informatique capable de « combler les images manquantes » du mouvement des protéines, même pour d’énormes machines moléculaires qui étaient auparavant trop grandes et complexes pour être simulées sur un ordinateur ordinaire.
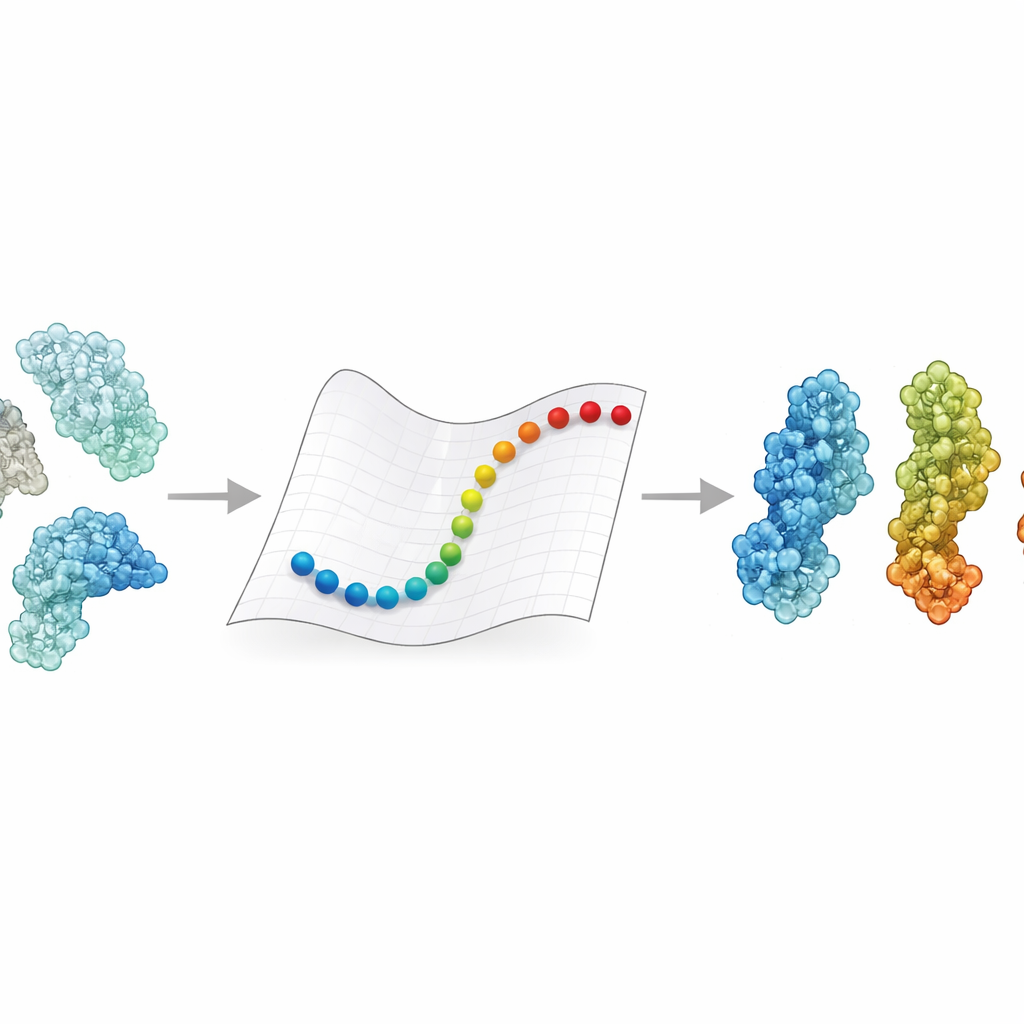
Pourquoi les changements de forme des protéines comptent
Les protéines restent rarement figées dans une seule posture. Elles s’ouvrent et se ferment, se tordent et se fléchissent en réponse à des signaux tels que des variations de voltage, de pH ou la liaison d’un partenaire moléculaire. Ces changements peuvent faire la différence entre une enzyme active ou inactive, ou entre un récepteur qui capture un virus ou le laisse échapper. Les expériences nous donnent des instantanés détaillés de quelques formes clés, et les simulations de dynamique moléculaire peuvent en principe les relier en suivant chaque atome au cours du temps. Mais suivre de tels mouvements pour les complexes gigantesques désormais observés par cryo‑EM, souvent de centaines de milliers à millions de daltons, exige généralement des supercalculateurs et des semaines de calcul. Par conséquent, pour de nombreux géants d’intérêt médical, nous ne savons toujours pas comment un état se transforme en un autre.
Un itinéraire plus rapide à travers les paysages protéiques
eBDIMS2 prend un raccourci en simplifiant la représentation des protéines et le calcul de leur mouvement. Plutôt que de suivre chaque atome, il traite chaque acide aminé comme un point unique relié par des ressorts dans un réseau élastique. Ces ressorts captent la tendance des différentes parties de la protéine à bouger ensemble. La méthode utilise ensuite la dynamique brownienne — des règles mathématiques qui imitent l’agitation dans un liquide — pour pousser la structure d’un état expérimental connu vers un autre. De manière cruciale, eBDIMS2 ne retient que les interactions réellement importantes, en appliquant des coupures de distance et en exploitant le calcul parallèle pour réduire le coût. Cela améliore l’évolutivité du programme, qui passe d’un comportement à peu près quadratique à presque linéaire par rapport à la taille de la protéine. En pratique, cela signifie que les transitions pour des assemblages énormes approchant deux millions de daltons peuvent être explorées en quelques heures sur un poste de travail, au lieu d’être essentiellement inaccessibles.
Comparer les trajectoires aux protéines réelles
Pour vérifier si ces trajectoires rapides ont un sens biologique, les auteurs ont constitué des ensembles de 47 grosses protéines et 15 complexes supplémentaires, totalisant des centaines de structures principalement résolues par cryo‑EM. Ils ont utilisé l’analyse en composantes principales, un outil statistique qui met en évidence les modes dominants de mouvement de chaque protéine, pour organiser ces structures en paysages de conformations tels qu’ouvert, fermé, actif ou inactif. eBDIMS2 a ensuite été chargé de connecter des paires d’états finals sur ce paysage. Les trajectoires obtenues ont été projetées sur les mêmes cartes de basse dimension, révélant si elles suivent des routes lisses passant près d’intermédiaires observés expérimentalement. Dans plus de 30 % des systèmes, les itinéraires simulés passaient à proximité — à quelques ångströms — d’intermédiaires qui n’avaient pas été fournis en entrée. Pour des cas exigeants comme l’enzyme de réparation de l’ADN DNA‑PKcs ou la protéine spike du coronavirus, les trajectoires en représentation grossière se recoupaient également bien avec des simulations atomiques beaucoup plus coûteuses, y compris la dynamique moléculaire ciblée et des méthodes avancées d’échantillonnage amélioré.
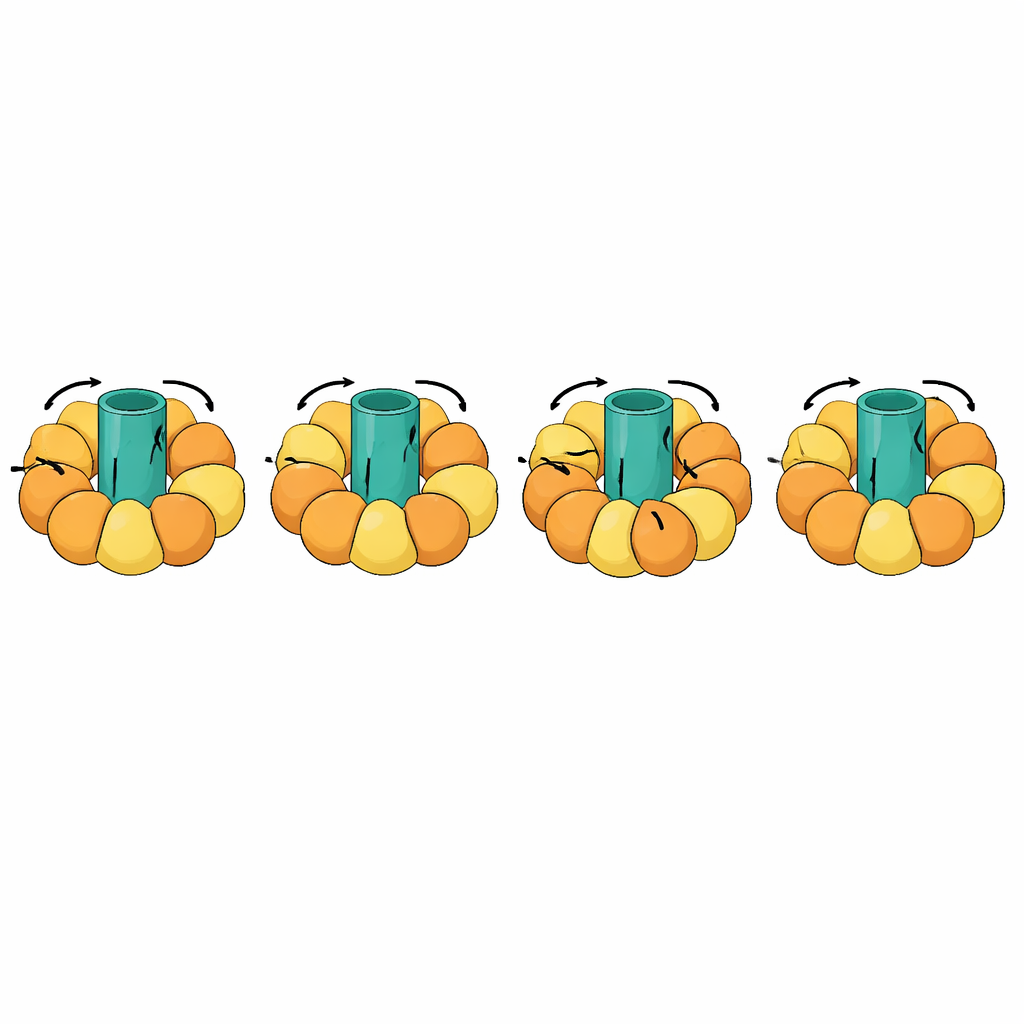
Suivre des machines moléculaires géantes
Un des tests les plus frappants concernait des machines rotatives telles que les ATP synthases, qui produisent la monnaie énergétique de la cellule en couplant un rotor tournant dans la membrane à des mouvements d’ouverture et de fermeture des sous‑unités environnantes. Ces transitions sont exceptionnellement complexes : certaines parties de la molécule doivent rester rigides et tourner comme une unité, tandis que d’autres se fléchissent dans un cycle chorégraphié. eBDIMS2 introduit une prise en charge spéciale pour de telles pièces quasi‑rigides et pour des modèles expérimentaux incomplets comportant des segments manquants, fréquents en cryo‑EM. Grâce à ces fonctionnalités, il peut simuler des cycles rotatifs complets d’ATP synthase et d’autres complexes massifs comme les chaperons moléculaires, les récepteurs et les assemblages viraux. Tout au long du processus, les structures intermédiaires générées évitent les distorsions sévères produites par certaines méthodes concurrentes et peuvent être affinées en modèles atomistiques adaptés aux calculs de conception de médicaments ou à des simulations plus longues et détaillées.
Ce que cela signifie pour la biologie et la médecine
L’étude montre qu’eBDIMS2 peut esquisser de façon fiable les principales voies entre formes protéiques connues pour des systèmes auparavant inaccessibles aux simulations traditionnelles. Il ne remplace pas des films atomiques détaillés ni ne fournit des énergies et des chronologies précises, mais il offre un moyen rapide et physiquement fondé de cartographier comment de grandes machines moléculaires pourraient se mouvoir, en n’utilisant qu’une paire de structures expérimentales en entrée. À mesure que les bases de données structurales se remplissent d’états multiples de grands assemblages protéiques liés au cancer, aux infections et à d’autres maladies, cette approche donne aux chercheurs un outil accessible pour relier les points, suggérer des états intermédiaires plausibles et orienter où chercher ensuite avec des méthodes à plus haute résolution ou des stratégies de conception de médicaments ciblées.
Citation: Scaramozzino, D., Lee, B.H. & Orellana, L. Efficient sampling of large-scale transition pathways and intermediate conformations in sub-mesoscopic protein complexes. Nat Commun 17, 2202 (2026). https://doi.org/10.1038/s41467-026-69809-y
Mots-clés: dynamique des protéines, simulations moléculaires, cryo‑EM, voies conformationnelles, modélisation grossière