Clear Sky Science · fr
Activation d’IRF3 dans les cardiomyocytes altère la fonction oxydative mitochondriale via l’inhibition de PGC-1α et entraîne une insuffisance cardiaque
Pourquoi les cœurs stressés et les cellules fatiguées comptent
L’insuffisance cardiaque est souvent décrite comme un « usure » du cœur, mais en coulisses c’est aussi une histoire d’inflammation chronique et de centrales énergétiques épuisées à l’intérieur des cellules musculaires cardiaques. Cette étude pose une question apparemment simple mais aux implications importantes : existe‑t‑il un commutateur moléculaire unique dans les cellules cardiaques qui relie l’inflammation nocive et la défaillance de la production d’énergie ? Et si oui, actionner ce commutateur peut‑il modifier la trajectoire de l’insuffisance cardiaque ? En suivant ce fil, les auteurs identifient un acteur clé et montrent qu’un renforcement modéré du programme énergétique propre au cœur peut partiellement sauver des cœurs en défaillance chez la souris.
Un commutateur moléculaire dans des cœurs humains malades
Les chercheurs se sont concentrés sur une protéine appelée IRF3, surtout connue pour son rôle dans la réponse aux infections virales. Ils ont examiné des tissus de personnes atteintes de cardiomyopathie ischémique, une forme courante d’insuffisance cardiaque due à une réduction du flux sanguin après un infarctus. Dans ces cœurs en échec, IRF3 n’était pas seulement présente : elle était chimiquement activée sur des sites spécifiques, signe qu’elle pilotait activement des programmes géniques. Parallèlement, la machinerie permettant aux mitochondries de convertir le carburant en énergie via la phosphorylation oxydative était visiblement affaiblie. Un schéma similaire est apparu dans des modèles murins d’infarctus : après la ligature d’une artère coronaire, IRF3 dans les cellules musculaires cardiaques s’activait fortement et les gènes régulés par IRF3 s’exprimaient. Même des fragments d’ADN mitochondrial — libérés par des mitochondries endommagées et agissant comme des signaux internes de danger — suffisaient à activer IRF3 dans des cellules cardiaques isolées.
Éteindre IRF3 protège le cœur
Pour tester si l’activité d’IRF3 dans les cellules cardiaques aggrave réellement la maladie, l’équipe a conçu des souris où IRF3 pouvait être supprimée uniquement dans les cardiomyocytes, sans toucher les autres cellules immunitaires ou de soutien. Après induction d’un infarctus, ces souris présentaient une fonction de pompage meilleure et moins de cicatrisation que les souris normales, malgré une lésion initiale équivalente. Dans des cellules cardiaques cultivées en lignée, le silence d’IRF3 réduisait l’expression des gènes inflammatoires sans perturber d’autres protéines apparentées. Dans l’ensemble, ces résultats soutiennent que IRF3, au sein même de la cellule cardiaque, n’est pas un simple témoin : elle amplifie l’inflammation et les lésions structurelles après l’ischémie et favorise la transition vers l’insuffisance cardiaque.
Quand IRF3 reste « allumé », le système énergétique s’effondre
Les auteurs ont ensuite inversé l’expérience : ils ont créé des souris dans lesquelles IRF3 dans les cardiomyocytes pouvait être maintenue en état d’activation permanente via une astuce génétique « phosphomimétique ». Sans stimulus externe, ces souris ont rapidement développé une dysfonction cardiaque sévère, des niveaux élevés de médiateurs inflammatoires dans le sang et des signes de lésion cellulaire. Une analyse approfondie de leur tissu cardiaque a montré que, sous activation chronique d’IRF3, elle supprime un coordinateur maître de l’énergie nommé PGC‑1α. Cette molécule favorise normalement des mitochondries saines, la combustion efficace des lipides et l’équilibre énergétique cellulaire. Lorsque PGC‑1α est diminué, de nombreuses protéines mitochondriales déclinent, la chaîne de transport d’électrons s’affaiblit et le choix des carburants du cœur se modifie : la carnitine et les composés associés à l’oxydation des graisses chutent, l’utilisation des corps cétoniques est altérée et le métabolisme du glucose se perturbe. Même le rapport NAD⁺/NADH — un indicateur clé de l’équilibre redox cellulaire — bascule dans une mauvaise direction.
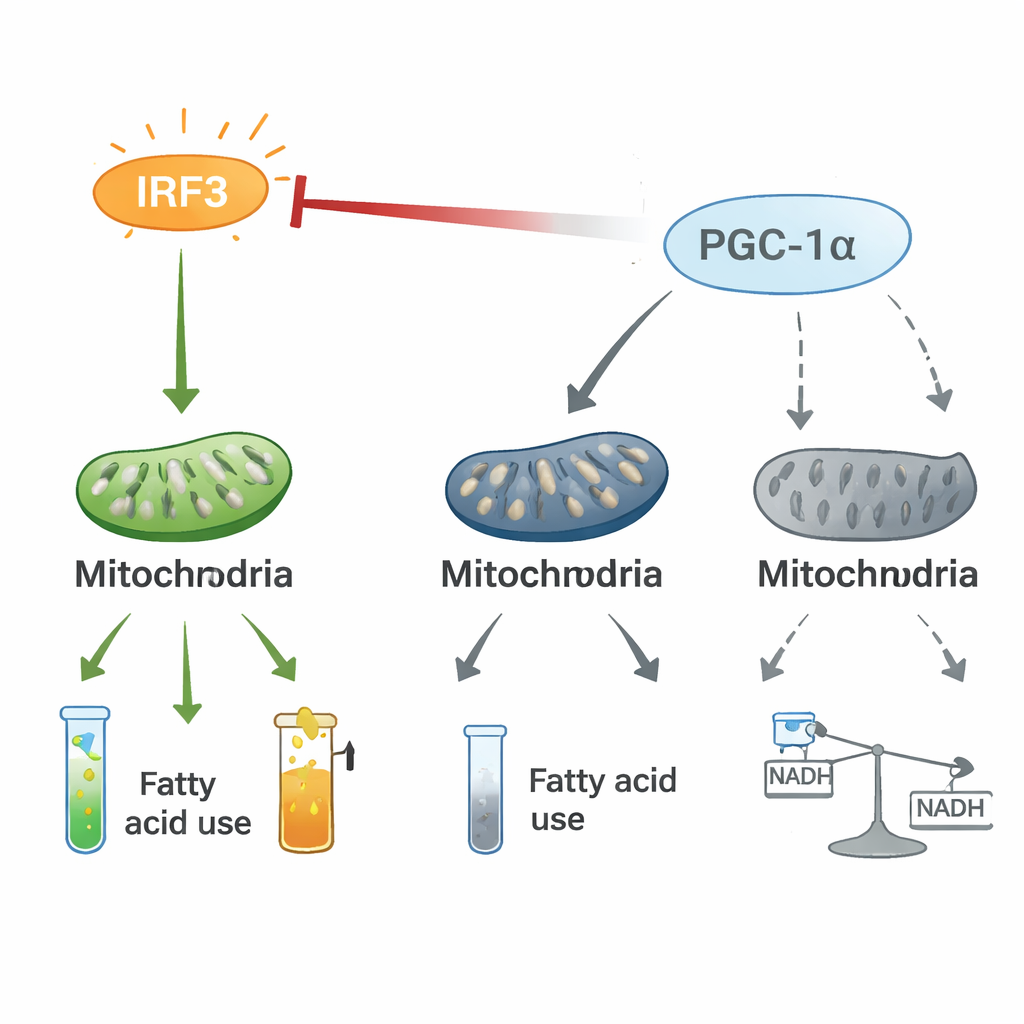
Un bras de fer entre inflammation et contrôle énergétique
Des expériences mécanistiques ont révélé qu’IRF3 et PGC‑1α forment un axe de régulation bidirectionnel. Dans les cellules cardiaques, IRF3 activée s’associe physiquement à PGC‑1α et atténue sa capacité à activer les gènes de la combustion des graisses. L’inhibition d’IRF3 élève les niveaux et l’activité de PGC‑1α, tandis que l’augmentation de PGC‑1α réduit l’expression des gènes inflammatoires pilotés par IRF3 et restaure les marqueurs mitochondriaux, y compris dans des conditions de stress comme l’hypoxie ou l’exposition à des toxines bactériennes. Le traçage par isotopes stables a montré que l’activation d’IRF3 réoriente le carbone, normalement destiné au cycle de l’acide citrique, vers une voie alternative — la voie des pentoses — et perturbe l’écoulement fluide des métabolites. Ce bras de fer entre un commutateur pro‑inflammatoire (IRF3) et un copilote énergétique (PGC‑1α) semble remodeler le métabolisme cardiaque de façon à favoriser l’inflammation et la perte d’énergie.
Recharger doucement les batteries du cœur
Enfin, les auteurs ont testé si remonter PGC‑1α pouvait contrer les effets délétères d’IRF3. Ils ont utilisé un vecteur de thérapie génique ciblant le cœur pour augmenter modérément — sans excès — PGC‑1α dans les mêmes souris présentant une IRF3 hyperactive. Cette augmentation modeste a amélioré la fonction de pompage, augmenté les protéines mitochondriales, renforcé l’expression des gènes de l’oxydation des lipides et du métabolisme du NAD, et réduit l’activité des gènes inflammatoires et fibrose. En culture cellulaire, l’expression conjointe de PGC‑1α avec IRF3 active a restauré un rapport NAD⁺/NADH plus sain et réorienté l’utilisation des carburants vers les lipides. Pour un lecteur non spécialiste, cela signifie que recharger prudemment le « système de gestion de batterie » du cœur peut partiellement compenser les effets nuisibles d’un commutateur inflammatoire chronique bloqué sur « marche ».
Ce que cela signifie pour la prise en charge future de l’insuffisance cardiaque
Ce travail positionne IRF3 comme un lien central entre inflammation et défaillance énergétique au sein des cellules musculaires cardiaques. Plutôt que de traiter l’inflammation et le métabolisme comme des problèmes séparés dans l’insuffisance cardiaque, l’étude suggère qu’ils sont intriqués via un axe IRF3–PGC‑1α. Bien que ces observations proviennent de souris et de modèles cellulaires, elles ouvrent la possibilité que des thérapies futures visent soit à réduire l’activité d’IRF3, soit à renforcer PGC‑1α et la fonction mitochondriale pour ralentir ou prévenir l’insuffisance cardiaque après un infarctus. En termes simples, calmer un système d’alarme cellulaire hyperactif et soutenir les usines énergétiques du cœur pourraient constituer une stratégie combinée puissante pour maintenir plus longtemps la performance des cœurs fragilisés.
Citation: Kumari, M., Evangelakos, I., Deshpande, A. et al. Activation of IRF3 in cardiomyocytes impairs mitochondrial oxidative function through PGC-1α inhibition and drives heart failure. Nat Commun 17, 2051 (2026). https://doi.org/10.1038/s41467-026-69792-4
Mots-clés: insuffisance cardiaque, inflammation, mitochondries, cardiomyocytes, PGC-1α