Clear Sky Science · fr
XL-MSDigger : une solution polyvalente basée sur l’apprentissage profond pour la spectrométrie de masse par réticulation
Voir comment les protéines tiennent ensemble
Tous les processus de notre organisme dépendent non seulement du repliement correct des protéines, mais aussi de leur capacité à trouver les bons partenaires. Pourtant, observer ces relations moléculaires en action est notoirement difficile. Cette étude présente XL-MSDigger, une plateforme logicielle qui utilise l’intelligence artificielle moderne pour extraire des signaux beaucoup plus clairs d’une technique expérimentale bruyante appelée spectrométrie de masse par réticulation, aidant les scientifiques à cartographier l’organisation des protéines et leurs interactions à l’intérieur des cellules.
Démêler un monde moléculaire encombré
Pour comprendre comment les protéines sont construites et comment elles se connectent, les chercheurs utilisent souvent la spectrométrie de masse par réticulation. Dans cette approche, de petits « ponts » chimiques lient entre elles des parties de protéines proches. Les fragments liés sont ensuite coupés en peptides et pesés dans un spectromètre de masse. En principe, le schéma des fragments révèle quelles parties de protéines étaient proches dans l’espace, comme découvrir quelles pages d’un livre avaient été agrafées ensemble. En pratique, cependant, les données obtenues sont extrêmement complexes. Les outils informatiques existants se contentent principalement des informations de masse de base et peinent à gérer l’énorme nombre de combinaisons possibles, entraînant des interactions manquées et des correspondances erronées.
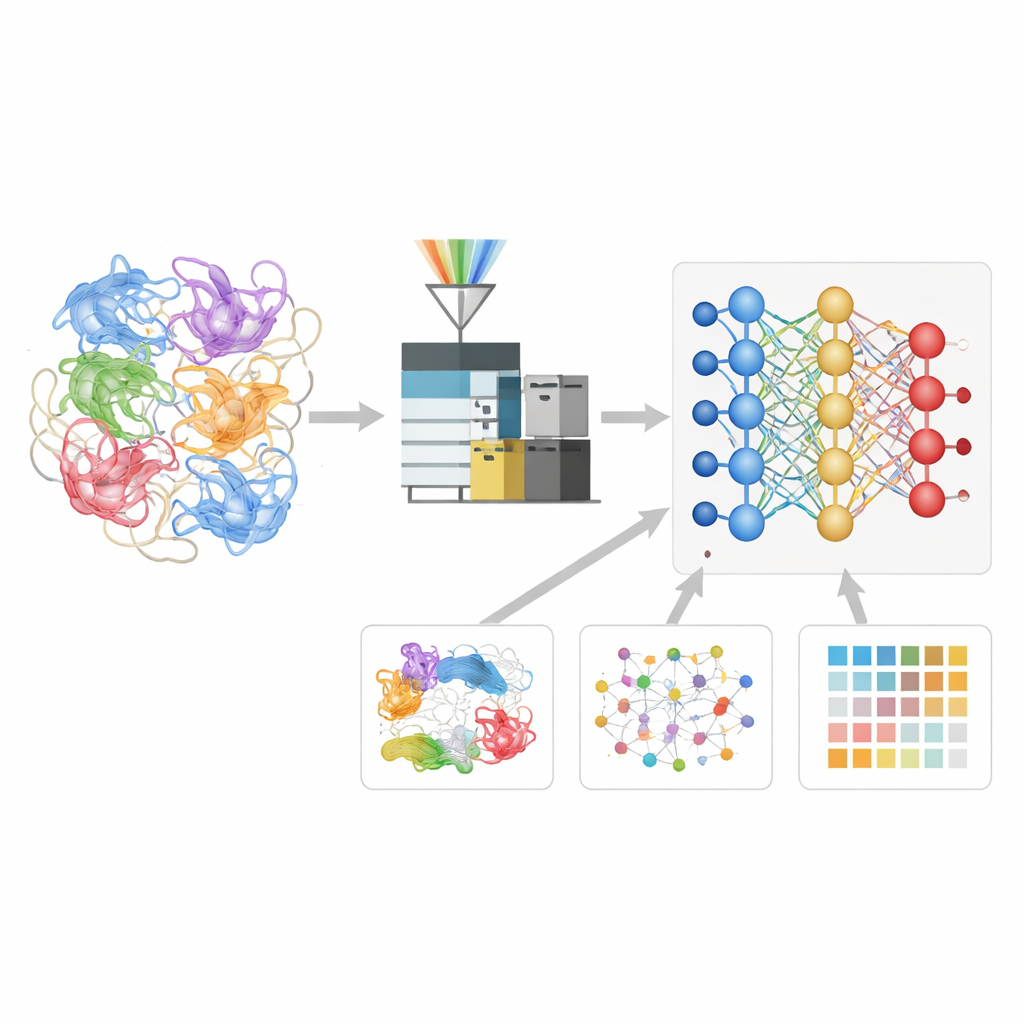
Apprendre au réseau de neurones le langage des fragments protéiques
Les auteurs ont construit un modèle d’apprentissage profond appelé Deep4D-XL pour mieux interpréter ces expériences de réticulation. Ils ont d’abord créé un grand jeu de référence en réticulant des protéines de cellules humaines, en les découpant en peptides et en enregistrant non seulement leurs masses, mais aussi leur temps de rétention dans l’instrument et leur comportement dans une chambre de mobilité ionique. Chaque paire réticulée a été encodée pour le modèle, qui utilise une architecture jumelle « Siamese » pour lire les deux peptides partenaires et une étape d’attention croisée pour combiner leurs informations. À partir de là, le réseau apprend à prédire trois propriétés clés de tout peptide réticulé nouveau : quand il doit apparaître dans l’expérience, comment il doit se comporter et à quoi doit ressembler son schéma de fragmentation.
Transformer les prédictions en signaux plus propres
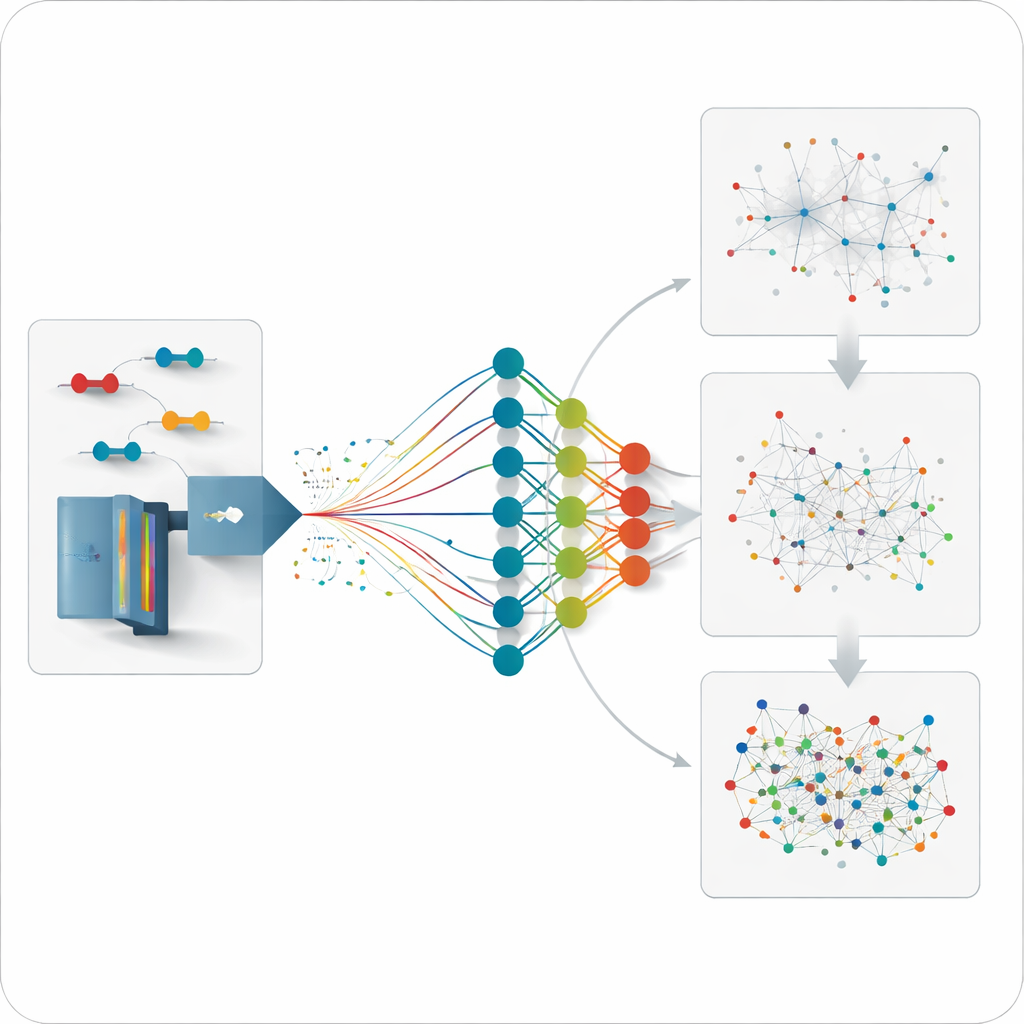
XL-MSDigger intègre ce moteur de prédiction dans des flux d’analyse pour deux principaux modes d’acquisition. Dans le mode traditionnel ciblé, l’instrument enregistre sélectivement les fragments d’ions qu’il choisit en temps réel. XL-MSDigger reprend les premières correspondances issues de logiciels de recherche établis et les réévalue en utilisant le comportement prédit par le modèle pour chaque candidat. Un second réseau de neurones compare prédiction et expérience selon plusieurs dimensions et attribue des scores améliorés. Cette réévaluation double presque le nombre de liaisons détectées avec confiance entre protéines différentes dans des échantillons de levure et humains tout en maintenant des taux d’erreur faibles, révélant de nombreuses interactions protéine–protéine supplémentaires par rapport à avant.
Rendre intelligible un flot de données non biaisées
Une manière plus récente d’exploiter ces instruments, appelée acquisition indépendante des données, enregistre les fragments pour presque tout ce qui se trouve dans un échantillon, améliorant la couverture mais générant des données écrasantes. Jusqu’à présent, il n’existait pas de bonne méthode pour estimer combien des réticulations résultantes étaient réellement réelles. XL-MSDigger utilise Deep4D-XL pour construire une bibliothèque « leurre » soigneusement appariée de réticulations factices, puis analyse entrées réelles et leurres ensemble. En observant la fréquence à laquelle les leurres passent le filtrage, le logiciel peut estimer le taux de fausses découvertes et entraîner un autre réseau de neurones à séparer les vraies correspondances des fausses. Cette réévaluation multiplie par environ cinq le nombre de signaux réticulés fiables et produit une séparation nette entre les schémas réels et ceux des leurres.
Prédire ce qui n’a pas encore été mesuré
Parce que le modèle peut prévoir le comportement de tout peptide réticulé plausible, l’équipe peut aller plus loin et analyser des données pour des liaisons qui n’ont jamais été mesurées directement. Ils génèrent des bibliothèques prédictives de taille modérée centrées sur des protéines sélectionnées ou des réseaux d’interaction, puis recherchent les données non biaisées contre ces bibliothèques. Cette stratégie révèle des liaisons supplémentaires au sein de protéines individuelles et entre partenaires de chaperonnes importantes, avec des distances en accord avec des structures tridimensionnelles connues. Elle récupère aussi des interactions manquées par les bibliothèques expérimentales traditionnelles, plus limitées, en particulier pour des liaisons de faible abondance.
Ouvrir une fenêtre plus claire sur les partenariats protéiques
Pour les non-spécialistes, le message principal est que XL-MSDigger agit comme un puissant reconnaisseur de motifs superposé à une méthode expérimentale déjà robuste. En apprenant à quoi doivent ressembler de véritables signaux réticulés sur plusieurs dimensions à la fois, il peut trier d’immenses jeux de données désordonnés, écarter les imposteurs probables et sauver des connexions protéiques réelles mais jusque-là cachées. Bien que des applications à l’échelle du protéome entier demanderont encore d’importantes ressources de calcul, ce travail montre que la combinaison d’expériences de réticulation et d’apprentissage profond peut considérablement affiner notre vision de l’organisation des protéines et de leurs rencontres à l’intérieur de la cellule.
Citation: Chen, M., Hao, Y., Huang, X. et al. XL-MSDigger: a deep learning-based, versatile solution for cross-linking mass spectrometry. Nat Commun 17, 2554 (2026). https://doi.org/10.1038/s41467-026-69489-8
Mots-clés: interactions protéiques, spectrométrie de masse par réticulation, apprentissage profond, protéomique, acquisition indépendante des données