Clear Sky Science · fr
Une référence générique définie par des peaks consensuels pour l’analyse des données ATAC-seq en cellules uniques
Pourquoi cartographier les portes ouvertes de notre ADN est important
Chaque cellule de votre corps porte essentiellement le même ADN, pourtant les cellules du cerveau, du sang ou les cellules tumorales se comportent très différemment. Une raison majeure est que seules certaines régions de l’ADN sont exposées et « ouvertes » à un moment donné. Les nouvelles technologies unicellulaires peuvent désormais mesurer cette ouverture à l’échelle du génome, mais jusqu’à présent elles manquaient d’une carte de référence commune — une sorte d’atlas standard — pour comparer les résultats entre expériences et laboratoires. Cette étude construit une telle carte, appelée cPeaks, et montre comment elle peut affiner notre compréhension des types cellulaires, du développement et du cancer.
Transformer de nombreuses expériences en une carte partagée
Les auteurs ont commencé par rassembler 624 expériences de haute qualité qui mesuraient la chromatine accessible — les parties accessibles de l’ADN — dans plus de 40 organes humains. Dans chaque expérience, des programmes informatiques avaient déjà détecté des « peaks » où l’ADN était particulièrement exposé. Plutôt que de traiter chaque jeu de données séparément, l’équipe a superposé avec soin toutes ces listes de peaks le long du génome et a fusionné les régions chevauchantes. Ils ont ensuite examiné la fréquence à laquelle chaque position minuscule à l’intérieur de ces régions fusionnées était appelée ouverte dans les expériences, transformant chaque région en une forme caractéristique reflétant sa constance d’apparition. Lorsqu’une région fusionnée contenait en réalité plusieurs sites ouverts rapprochés, ils l’ont scindée en plusieurs unités plus simples. Ces unités — environ 1,4 million au total — sont devenues les peaks consensuels observés, ou cPeaks, un catalogue de référence candidat pour l’accessibilité de la chromatine humaine. 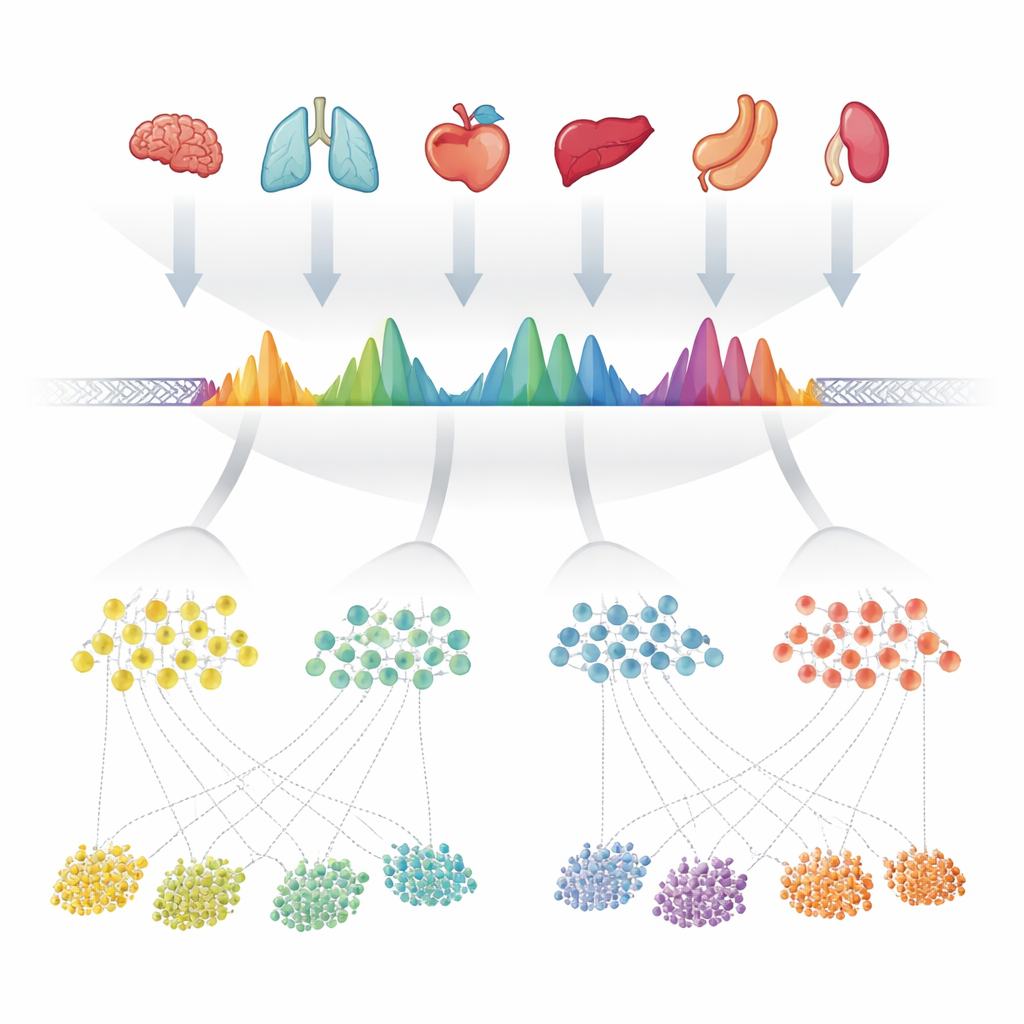
Une empreinte stable à travers tissus et technologies
Pour être une référence utile, ces cPeaks doivent représenter des caractéristiques véritables et répétables du génome, et non des particularités d’échantillons ou de logiciels. Les auteurs ont testé cela en recréant leurs régions fusionnées en n’utilisant que des échantillons sanguins, seulement des tissus solides, des bases de données publiques séparées, et même différentes méthodes de laboratoire pour sonder l’ADN ouvert. Dans chaque cas, les mêmes emplacements génomiques produisaient des formes de peaks remarquablement similaires, et la plupart des jeux de données unicellulaires examinés chevauchaient plus de 90 % de leurs propres peaks avec le catalogue cPeak. Les lectures provenant de nombreux organes s’accumulaient précisément autour des centres de cPeaks, montrant que ces régions capturent de manière fiable où la chromatine est ouverte. Par rapport aux ensembles de référence précédents basés sur des technologies apparentées, cPeaks couvraient une plus grande partie de l’ADN accessible détecté par les expériences ATAC-seq, et capturaient presque autant de signal que des peaks définis ex nihilo dans chaque jeu de données — malgré le fait d’être fixes et réutilisables.
Apprendre à un réseau de neurones à retrouver des régions manquantes
Même des centaines d’échantillons existants ne peuvent couvrir tous les types cellulaires possibles. Pour étendre leur carte vers des régions encore non observées, l’équipe a recours à l’apprentissage profond. Ils ont entraîné un réseau neuronal convolutionnel unidimensionnel sur des séquences d’ADN : des exemples situés à l’intérieur des cPeaks observés servaient de positifs, tandis que des régions de fond choisies aléatoirement servaient de négatifs. Le modèle a appris à distinguer ces deux classes avec une grande précision, impliquant que les cPeaks portent des motifs de séquence reconnaissables. Lorsque les chercheurs ont délibérément caché les peaks spécifiques à un tissu à la fois, le réseau a tout de même pu les retrouver à partir de la séquence seule, y compris des sites rares et spécifiques à un tissu. Ils ont ensuite glissé une petite fenêtre le long du reste du génome, notant chaque segment et ajoutant environ 280 000 nouvelles régions à fort score au catalogue comme cPeaks prédites, améliorant particulièrement la couverture dans les tissus sous-représentés dans les données initiales.
Relier les régions ouvertes aux gènes, types cellulaires et cellules rares
Avec une référence enrichie en main, les auteurs ont cherché à savoir ce que font ces régions. De nombreux cPeaks se situent près des sites de début et de fin des gènes ou chevauchent des éléments régulateurs connus tels que promoteurs, enhancers et sites de liaison pour des protéines architecturales comme CTCF. Un petit sous-ensemble est accessible dans presque tous les jeux de données ; ces cPeaks « ménagères » plus longues tendent à se situer dans les régions promotrices centrales de gènes nécessaires au maintien cellulaire de base. L’équipe a également classé les cPeaks selon la netteté et la cohérence de leurs bords à travers les échantillons, ce qui reflète la précision de l’emballage de l’ADN voisin en nucléosomes. Les régions aux frontières bien définies sont enrichies pour certaines familles de facteurs de transcription connues pour remodeler la chromatine et piloter le développement. Quand les cPeaks ont été utilisés comme jeu de caractéristiques pour analyser plusieurs jeux de données unicellulaires, ils ont amélioré la précision de l’étiquetage des types cellulaires, et ont été particulièrement utiles pour identifier des types cellulaires rares et des sous-types subtils que les ensembles de peaks précédents ou des grilles génomiques simples ont souvent confondus.
Suivre le développement et le cancer avec un langage commun
La puissance d’une référence standard devient évidente lorsqu’on compare des contextes biologiques très différents. En utilisant cPeaks, les auteurs ont réanalysé des données unicellulaires de la rétine humaine en développement, de grands atlas de tissus fœtaux et adultes, et plusieurs cancers. Ils ont pu reconstruire des trajectoires développementales et observer que la fraction de cPeaks « bien positionnés » à bords nets tend à augmenter durant les stades transitionnels, puis à diminuer à mesure que les cellules se stabilisent dans des identités définies. Un schéma similaire est apparu à travers les stades tumoraux : les cancers intermédiaires présentaient une proportion plus élevée de ces régions structurées, suggérant un remodelage régulateur intense. Dans une tumeur ovarienne, les cPeaks ont aidé à révéler deux sous-clones tumoraux distincts avec des altérations différentes du nombre de copies d’ADN, montrant comment la référence peut exposer une complexité cachée dans la maladie.
Ce que cela signifie pour la recherche génomique future
Pour les non-spécialistes, les cPeaks peuvent être considérés comme un jeu standardisé de coordonnées indiquant où le génome est le plus susceptible d’être physiquement ouvert et actif à travers de nombreux types cellulaires humains. En alignant de nouvelles expériences unicellulaires de chromatine sur cette carte partagée, les chercheurs peuvent comparer les résultats entre études, repérer plus facilement des états cellulaires rares ou transitionnels, et commencer à construire des modèles à grande échelle de la régulation génique — de la même façon que les catalogues standardisés de gènes ont permis l’essor des atlas unicellulaires transcriptomiques. Le catalogue cPeak actuel est un premier brouillon qui grandira au fil des nouvelles données, mais il fournit déjà un langage commun pour décrire l’accessibilité de la chromatine, nous rapprochant d’une vue unifiée de la façon dont l’emballage de l’ADN guide le développement, la santé et la maladie. 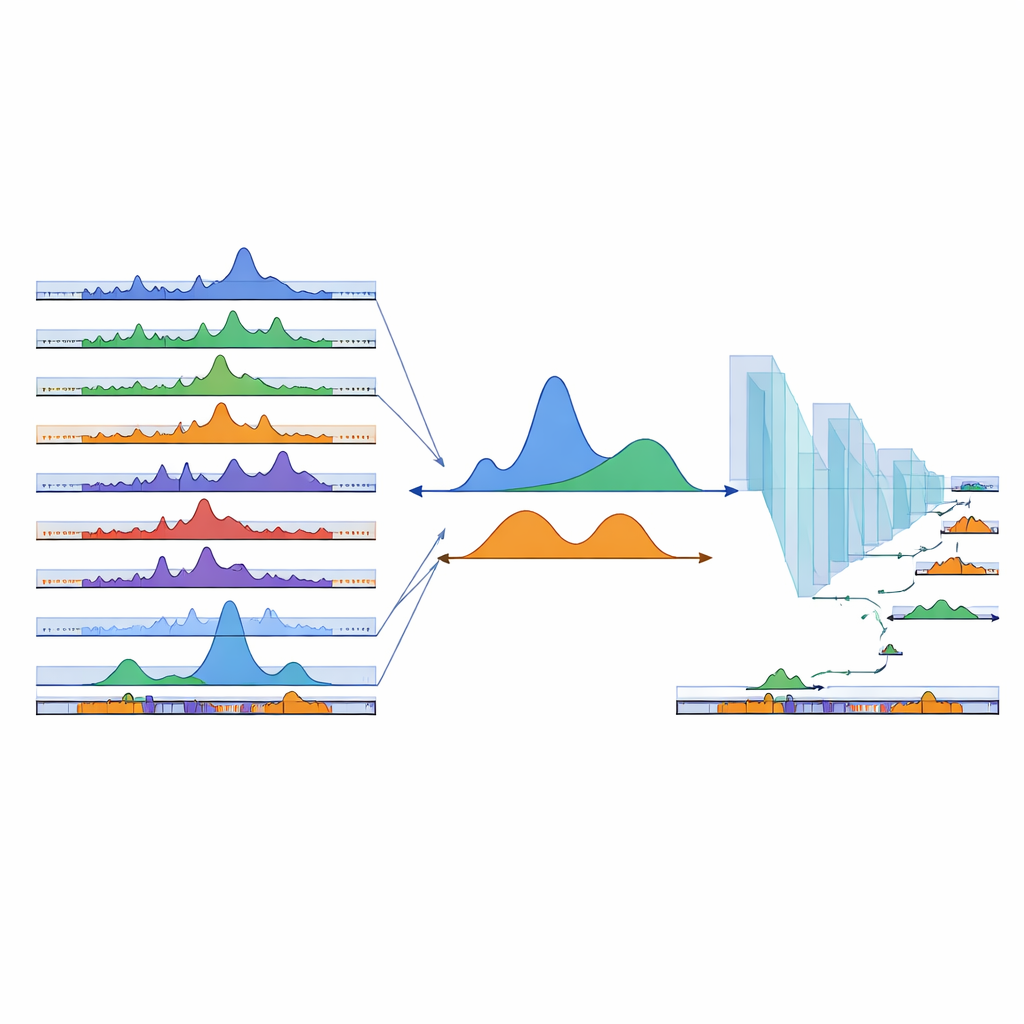
Citation: Meng, Q., Wu, X., Chen, W. et al. A generic reference defined by consensus peaks for single-cell ATAC-seq data analysis. Nat Commun 17, 2522 (2026). https://doi.org/10.1038/s41467-026-69461-6
Mots-clés: accessibilité de la chromatine, ATAC-seq unicellulaire, peaks consensuels, régulation génique, génomique et apprentissage profond