Clear Sky Science · fr
Séquençage de la méthylation et de l’hydroxyméthylation de l’ADN aux caractéristiques chromatiniennes co‑existantes
Lire les notes chimiques de nos cellules
Toutes les cellules du corps portent le même ADN, pourtant les cellules cérébrales, cutanées et souches se comportent très différemment. Une des raisons est que les cellules écrivent des « notes » chimiques sur l’ADN et sur les protéines qui l’enveloppent, aidant à activer ou à réprimer des gènes. Jusqu’à présent, les scientifiques ont eu du mal à lire simultanément plusieurs de ces notes sur une même portion d’ADN, laissant un vide dans notre compréhension de leur coopération. Cette étude présente une nouvelle méthode permettant de lire à la fois le code génétique et des marquages chimiques clés, révélant comment ils se combinent pour contrôler des interrupteurs d’ADN importants appelés enhancers.
Pourquoi l’ADN a besoin de marques au crayon
L’ADN n’agit pas seul. Il est enroulé autour de protéines appelées histones pour former la chromatine, et l’ADN comme les histones peuvent être ornés de petits groupes chimiques. Deux marques importantes sur l’ADN sont des groupes méthyl et hydroxyméthyl ajoutés à la lettre C (cytosine). Ces marques influencent la compaction de l’ADN et l’activité des gènes voisins. De manière générale, les marques méthyl sont souvent associées à la répression génique, tandis que les hydroxyméthyl ont tendance à apparaître là où les gènes sont actifs. Mais l’impact de ces marques dépend de leur contexte local : exactement où elles se situent dans le génome et à quels marquages d’histones elles sont juxtaposées.
Le problème des cartes séparées
Les méthodes de séquençage existantes peuvent cartographier la méthylation et l’hydroxyméthylation sur l’ensemble du génome, et d’autres méthodes cartographient les marques d’histones indiquant des régions actives ou silencieuses. Toutefois, ces expériences sont généralement réalisées séparément puis comparées par informatique. Cela indique quelles caractéristiques tendent à se trouver dans le même voisinage, mais pas si elles coexistent réellement sur le même fragment d’ADN dans une seule cellule. Les tentatives antérieures pour combiner ces mesures reposaient sur des traitements chimiques agressifs qui endommagent l’ADN et, surtout, ne permettaient pas de distinguer de façon fiable méthylation et hydroxyméthylation sur une même lecture. En conséquence, les chercheurs manquaient d’une image moléculaire claire de la coopération entre marques.
Une nouvelle méthode de lecture multi‑couche
Les auteurs ont mis au point une méthode appelée 6-base-CUT&Tag capable de lire les quatre lettres de l’ADN plus deux états chimiques de la cytosine — non modifiée, méthylée et hydroxyméthylée — sur des fragments d’ADN physiquement attachés à des caractéristiques chromatiniennes choisies. D’abord, ils utilisent des anticorps comme des crochets moléculaires pour extraire l’ADN enroulé autour d’histones portant une marque spécifique, par exemple un signe de chromatine active. Une enzyme conçue insère ensuite des adaptateurs spéciaux, transformant chaque fragment capturé en une petite boucle qui résiste aux étapes de purification qui détruisent les fragments non désirés. Un procédé chimique et enzymatique affiné convertit ensuite les différents états de la cytosine en signaux de séquence distincts, lisibles par les séquenceurs modernes. De cette façon, une seule lecture renseigne sur l’origine du fragment, la marque d’histone qu’il portait et quelles cytosines y étaient méthylées ou hydroxyméthylées.
Zoom sur les interrupteurs géniques
En utilisant des cellules souches embryonnaires de souris comme cas test, l’équipe a appliqué le 6-base-CUT&Tag à plusieurs marques d’histones clés qui signalent différents types d’ADN régulateur. Ils se sont concentrés sur les enhancers — des segments d’ADN qui fonctionnent comme des interrupteurs contrôlant le moment et le lieu d’activation des gènes. Les enhancers peuvent être « actifs », « amorcés » (primed) ou « en attente » (poised), distingués par des marques d’histones particulières. Les chercheurs ont constaté que les enhancers marqués uniquement par une balise d’histone appelée H3K4me1 (souvent considérée comme « amorcée ») portaient les niveaux les plus élevés de méthylation et d’hydroxyméthylation sur l’ADN, en particulier lorsqu’ils étaient examinés directement au niveau des nucléosomes liés à H3K4me1. En revanche, les enhancers présentant des signes supplémentaires d’activité forte ou de répression montraient moins de ces marques d’ADN ou une bascule vers l’hydroxyméthylation, suggérant une éradication en cours des marques méthylées.
Décoder les états d’enhancer avec plus de précision
Parce que tous les types d’enhancers partagent la marque H3K4me1, l’équipe a demandé si le motif détaillé des marques d’ADN spécifiquement au niveau de l’ADN étiqueté H3K4me1 pouvait à lui seul distinguer les différents états d’enhancer. Ils ont entraîné un modèle d’apprentissage automatique utilisant les données 6-base-CUT&Tag pour classer les enhancers en actifs, amorcés ou en attente, uniquement d’après la quantité de méthylation et d’hydroxyméthylation qu’ils portaient à ce seul site d’histone. Ce modèle a surpassé un modèle par ailleurs identique entraîné sur des données standard à l’échelle du génome non restreintes à une marque d’histone. Autrement dit, lire les marques d’ADN dans leur contexte immédiat offre une image plus nette que la moyenne calculée sur tout l’ADN de la cellule.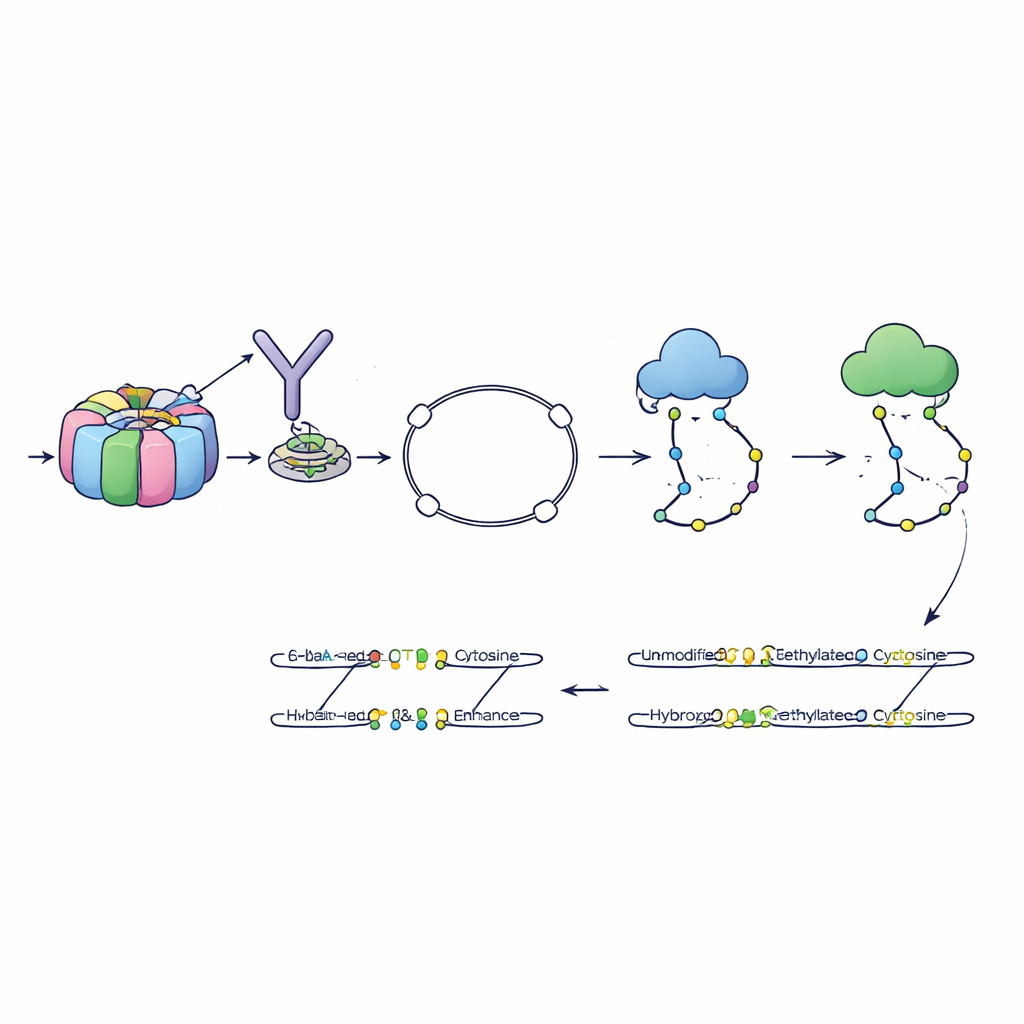
Ce que cela signifie pour la compréhension de l’identité cellulaire
Pour un non‑spécialiste, le message clé est que cette méthode permet aux scientifiques de lire plusieurs couches d’information — séquence d’ADN, marques d’ADN et marques d’histones — sur une même molécule. Cette vue fine révèle comment des combinaisons particulières d’étiquettes chimiques définissent la préparation des interrupteurs géniques dans les cellules souches. Parce que le 6-base-CUT&Tag est plus efficace et moins destructeur que les approches antérieures, il peut mettre en évidence des motifs subtils auparavant cachés. À terme, cette lecture multi‑couche de la chromatine pourrait aider à expliquer comment les cellules se souviennent de leur identité, comment elles changent pendant le développement ou la maladie, et comment l’on pourrait cibler plus précisément le code régulateur en thérapie.
Citation: Araujo Tavares, R.d.C., Dhir, S., He, X. et al. Sequencing DNA methylation and hydroxymethylation at co-occurring chromatin features. Nat Commun 17, 2591 (2026). https://doi.org/10.1038/s41467-026-69429-6
Mots-clés: épigénétique, méthylation de l’ADN, chromatine, enhancers, cellules souches