Clear Sky Science · fr
Des variants perte-de-fonction de l’activateur CAPN1 CD99L2 provoquent une ataxie spastique liée à l’X
Pourquoi cela compte pour les familles confrontées à des troubles du mouvement inexpliqués
De nombreuses personnes vivent pendant des années avec des difficultés de marche, une raideur musculaire ou des problèmes d’équilibre et de parole sans jamais connaître la véritable cause. Cette étude montre comment les tests ADN modernes peuvent enfin apporter des réponses à certaines de ces familles. Les chercheurs ont non seulement comparé différents tests génétiques pour les troubles rares du mouvement, mais ont aussi identifié une cause jusque-là inconnue d’une affection appelée ataxie spastique liée à l’X, mettant en lumière des voies biologiques qui pourraient également jouer un rôle dans des maladies cérébrales plus fréquentes.
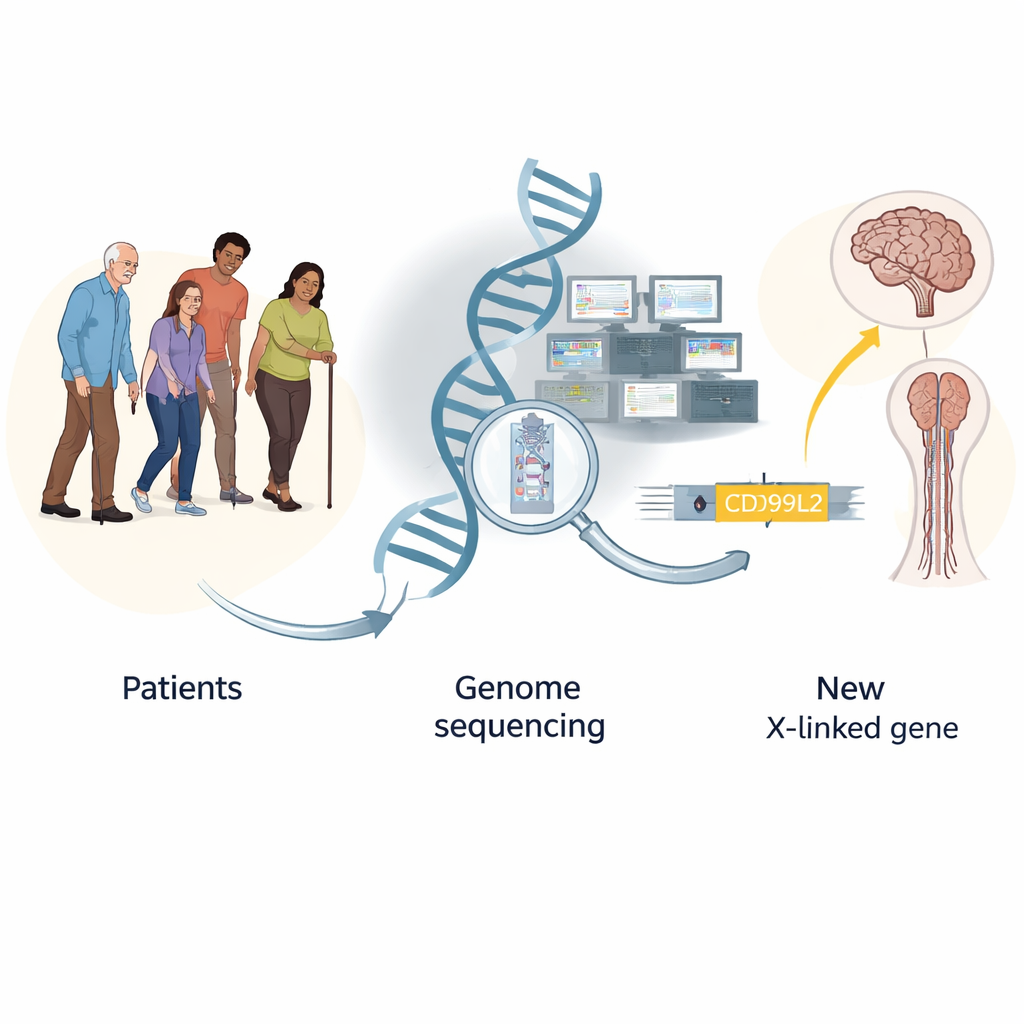
Retrouver les aiguilles génétiques dans la botte de foin des maladies rares
Les troubles rares du mouvement, comme l’ataxie (mouvements instables) et la paraplégie spastique (jambes raides et faibles), sont souvent suspectés d’origine génétique, mais pour la plupart des patients les tests standard restent négatifs. L’équipe a suivi 2 811 personnes en Allemagne et dans toute l’Europe, adressées pour suspicion de troubles rares du mouvement sur six ans. D’abord, ils ont réalisé des tests ciblés traditionnels qui recherchent des expansions de répétitions connues dans quelques gènes ; ces tests ont apporté une réponse dans environ 11 % des cas. Ensuite, le séquençage de l’exome, qui lit uniquement les parties codantes du génome, a fourni des explications génétiques définitives chez environ 19 % des patients, notamment chez ceux présentant de la spasticité.
Aller au‑delà des tests standards avec le séquençage du génome entier
Pour aller plus loin, les scientifiques ont utilisé le séquençage du génome entier, qui lit presque la totalité de l’ADN d’une personne, y compris des régions que les tests standard et l’exome peuvent manquer. Parmi 486 individus ayant bénéficié de ce test plus complet, le taux de diagnostic a augmenté d’environ 7,5 points de pourcentage, principalement parce que le séquençage du génome détecte mieux des altérations complexes telles que des réarrangements structuraux et des expansions de répétitions. L’étude montre également que des informations cliniques soigneusement consignées — en particulier des descriptions précises des symptômes, un âge plus jeune au moment du test et la combinaison de spasticité avec d’autres troubles du mouvement — ont aidé à prédire qui était le plus susceptible d’obtenir un diagnostic génétique clair.
Découverte d’une nouvelle cause liée à l’X de l’ataxie spastique
Même après ces examens approfondis, de nombreux patients restaient sans diagnostic. Les chercheurs ont regroupé des données génétiques de plus de 13 000 personnes et utilisé une approche de « charge génétique », qui consiste à rechercher les gènes portant des variants suspects plus souvent chez les malades que chez des témoins sains. Cette analyse a souligné non seulement des gènes déjà connus, mais a aussi fortement mis en évidence un gène jusque‑là négligé sur le chromosome X, appelé CD99L2. En combinant les résultats de plusieurs familles européennes, ils ont identifié 25 hommes atteints issus de 20 familles portant des variants délétères dans ce gène. Ces hommes développaient typiquement des troubles de la marche, une raideur des jambes, une élocution pâteuse et parfois des difficultés d’équilibre à l’âge moyen ou avancé, tandis que les femmes porteuses étaient pour la plupart indemnes — des schémas compatibles avec une maladie liée à l’X. Les variants détruisaient principalement la protéine normale ou éliminaient des régions cruciales, ce qui suggère fortement qu’une perte de fonction est à l’origine de la maladie.
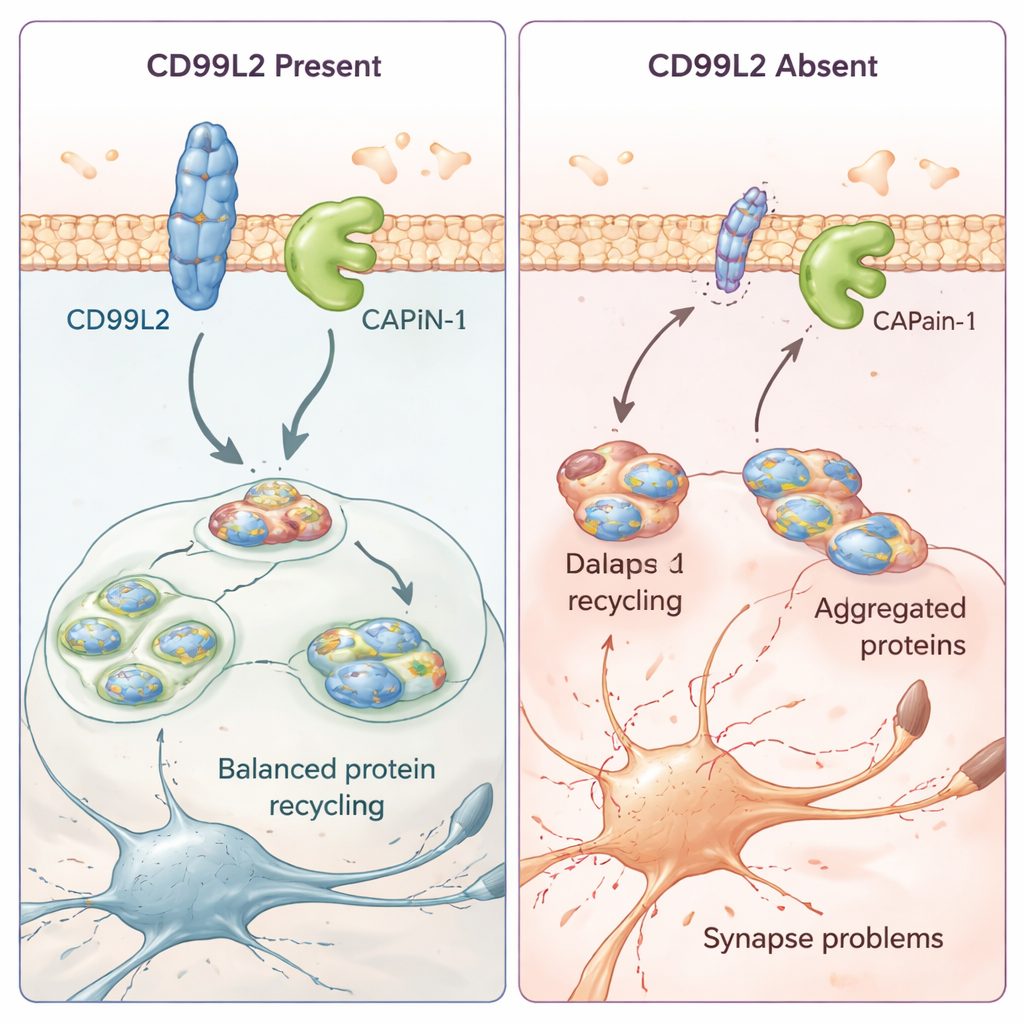
Comment une petite protéine membranaire protège les cellules cérébrales
Pour comprendre le rôle de CD99L2 dans les cellules, l’équipe a eu recours à des modèles cellulaires et à des cellules cutanées dérivées de patients. Ils ont découvert que la protéine CD99L2 se situe dans la membrane cellulaire et est généralement marquée par de petites étiquettes « ubiquitine » qui contrôlent sa durée de vie avant dégradation. CD99L2 se lie physiquement à la calpaïne‑1 (CAPN1), une enzyme activée par le calcium qui coupe d’autres protéines et contribue à maintenir la santé des synapses — les points de contact entre les cellules nerveuses. Lorsque CD99L2 est présente et intacte, elle aide à activer et désactiver la calpaïne‑1 de manière contrôlée, puis elle-même est clivée et recyclée. En l’absence de CD99L2 ou lorsque sa structure est altérée, l’activation de la calpaïne‑1 est perturbée. Dans les cellules de patients, cela s’accompagne d’une activité dérégulée de nombreux gènes liés aux synapses et à la communication neuronale, ce qui suggère que des modifications subtiles mais étendues du câblage cérébral pourraient sous‑tendre l’apparition progressive des troubles du mouvement.
Ce que cela signifie pour les patients aujourd’hui et demain
Pour les familles confrontées à une ataxie spastique ou une paraplégie spastique inexpliquées, ce travail apporte deux avancées. D’une part, il montre que le recours précoce au séquençage du génome entier, associé à une description clinique rigoureuse, peut augmenter de manière notable les chances d’obtenir un diagnostic génétique fiable. D’autre part, il ajoute CD99L2 à la liste des gènes qui régulent l’activité des calpaïnes, une voie déjà impliquée dans d’autres ataxies rares et dans des maladies courantes comme Alzheimer et Parkinson. Concrètement, l’étude révèle un nouveau « interrupteur » on‑off qui aide à maintenir l’équilibre de l’entretien des cellules nerveuses ; lorsque cet interrupteur est rompu, les neurones se détériorent lentement, entraînant raideur et mauvaise coordination. Mieux comprendre cet interrupteur pourrait, à terme, ouvrir la voie à des traitements visant à ajuster l’activité des calpaïnes et à protéger les cellules cérébrales dans une gamme de maladies neurologiques.
Citation: Menden, B., Incebacak Eltemur, R.D., Demidov, G. et al. Loss-of-function variants in the CAPN1 activator CD99L2 cause X-linked spastic ataxia. Nat Commun 17, 1698 (2026). https://doi.org/10.1038/s41467-026-69337-9
Mots-clés: ataxie spastique, troubles du mouvement rares, séquençage du génome, CD99L2, calpaïne‑1