Clear Sky Science · fr
Les états excités ferrique‑oxyle peuvent‑ils expliquer les liaisons fer‑oxygène allongées dans les intermédiaires catalytiques des peroxydases hémiques ?
Pourquoi les liaisons fer‑oxygène dans les enzymes sont importantes
À l’intérieur de nos cellules, des protéines spécialisées appelées enzymes utilisent l’oxygène pour réaliser en toute sécurité des réactions chimiques puissantes. Parmi elles, les peroxydases hémiques s’appuient sur une paire fer–oxygène en leur cœur pour décomposer le peroxyde d’hydrogène, une molécule réactive et potentiellement dommageable. Pendant des décennies, les scientifiques ont débattu de la nature exacte de cette liaison fer‑oxygène : s’apparente‑t‑elle davantage à une double liaison serrée ou à une liaison simple plus lâche — et qu’est‑ce que cela implique pour le fonctionnement des enzymes ? Cette étude aborde ce mystère en combinant méthodes X ultrarapides et calculs avancés, révélant que la réponse se trouve dans des états excités fugaces de l’unité fer‑oxygène elle‑même.
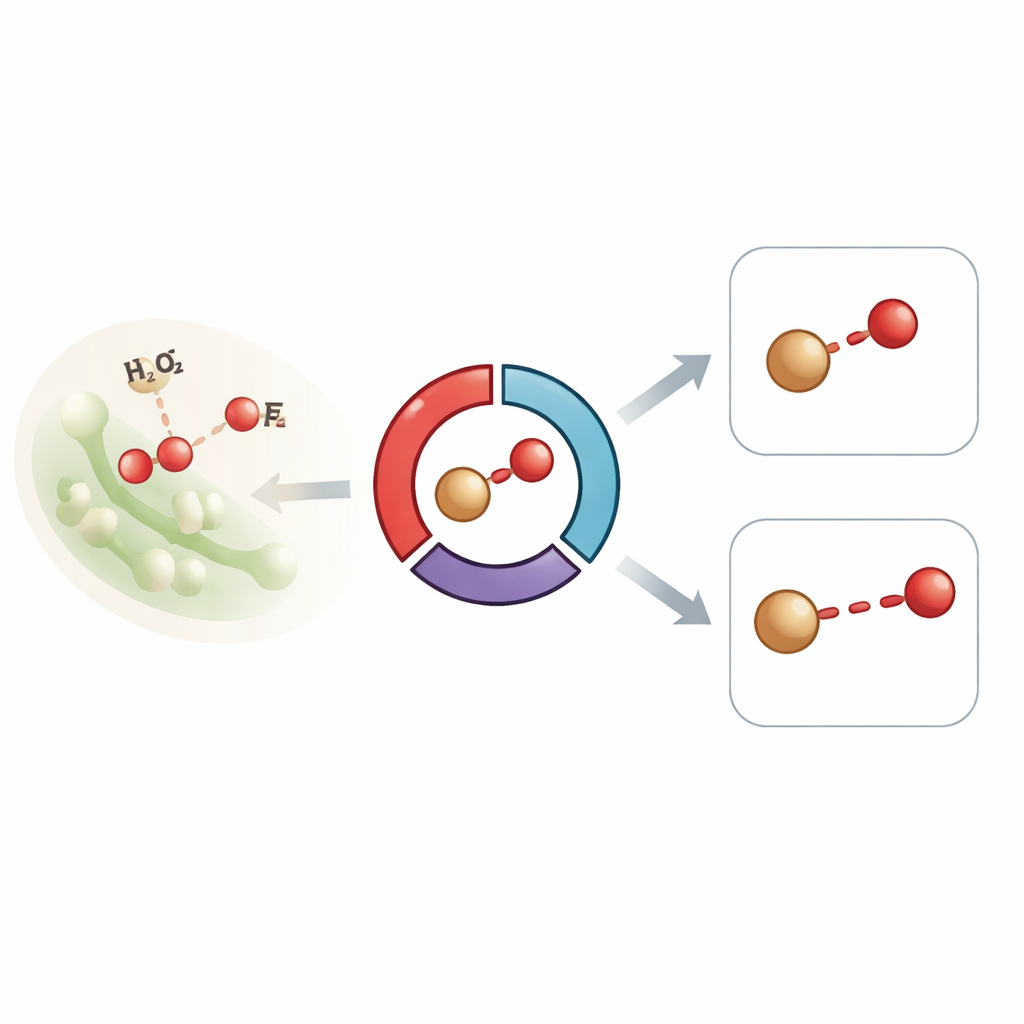
Suivre une enzyme en temps réel
Les chercheurs se sont concentrés sur une peroxydase bactérienne décolorante, une enzyme héminique qui évolue normalement entre deux formes énergétiques clés, connues sous les noms de Compound I et Compound II. Ces formes présentent toutes deux un atome de fer lié à l’oxygène et sont centrales pour la manière dont l’enzyme traite le peroxyde d’hydrogène et oxyde d’autres molécules. Des expériences antérieures sur des enzymes similaires avaient donné des longueurs de liaison fer‑oxygène étonnamment longues, que certains ont interprétées comme la preuve que l’unité fer‑oxygène hautement réactive avait été modifiée par les rayons X ou avait gagné un proton supplémentaire, changeant ainsi son caractère. Pour éviter de tels artefacts, l’équipe a utilisé la cristallographie sérielle femtoseconde résolue dans le temps à un laser à électrons libres X, enregistrant la diffraction et les signaux d’émission X à partir de milliers de microcristaux protéiques à température ambiante, le tout en quelques dizaines de femtosecondes — plus rapide que l’apparition des dommages.
Observer la chimie se dérouler dans les cristaux
Dans leur dispositif, des microcristaux d’une version légèrement modifiée de l’enzyme ont été mélangés au peroxyde d’hydrogène directement sur une bande mobile puis sondés après des délais allant d’une demi‑seconde à plusieurs dizaines de minutes. Les premiers instants favorisent la formation du Compound I, tandis que les temps plus longs sont dominés par le Compound II. Les données structurales ont montré que, dans les deux intermédiaires, l’atome de fer se situe à côté d’un seul atome d’oxygène dans la poche héminique, et que des régions en boucle protectrices de la protéine se déplacent pour isoler ce centre fortement oxydant. De manière importante, des mesures précises ont révélé que la longueur de la liaison fer‑oxygène restait proche de 1,83 angström à tous les instants — plus longue que prévu pour une espèce ferryl classique (Fe(IV)=O) et plus proche d’une liaison simple — pourtant les signatures spectrales d’émission X et d’absorption optique indiquaient clairement des états d’oxydation élevés compatibles avec les Compounds I et II.
Écarter les explications simples
Étant donné que les expériences ont été réalisées avec des impulsions ultracourtes à température ambiante, les causes habituelles de l’allongement apparent des liaisons — réduction induite par les rayons X et artefacts cryogéniques — peuvent être largement écartées. L’équipe a aussi testé si l’oxygène lié au fer avait été protoné, transformant une double liaison en une liaison simple de type hydroxyle. Cependant, les propriétés acido‑basiques connues de centres héminiques similaires, ainsi que des études chimiques antérieures, militent fortement contre une telle protonation dans ce type d’enzyme. Les données spectroscopiques ont en outre montré que le fer restait dans un état d’oxydation élevé et de bas spin après réaction avec le peroxyde d’hydrogène, comme attendu pour de véritables intermédiaires ferryl, renforçant l’idée que la liaison anormalement longue résulte d’effets électroniques plus subtils plutôt que d’un simple changement de forme chimique.
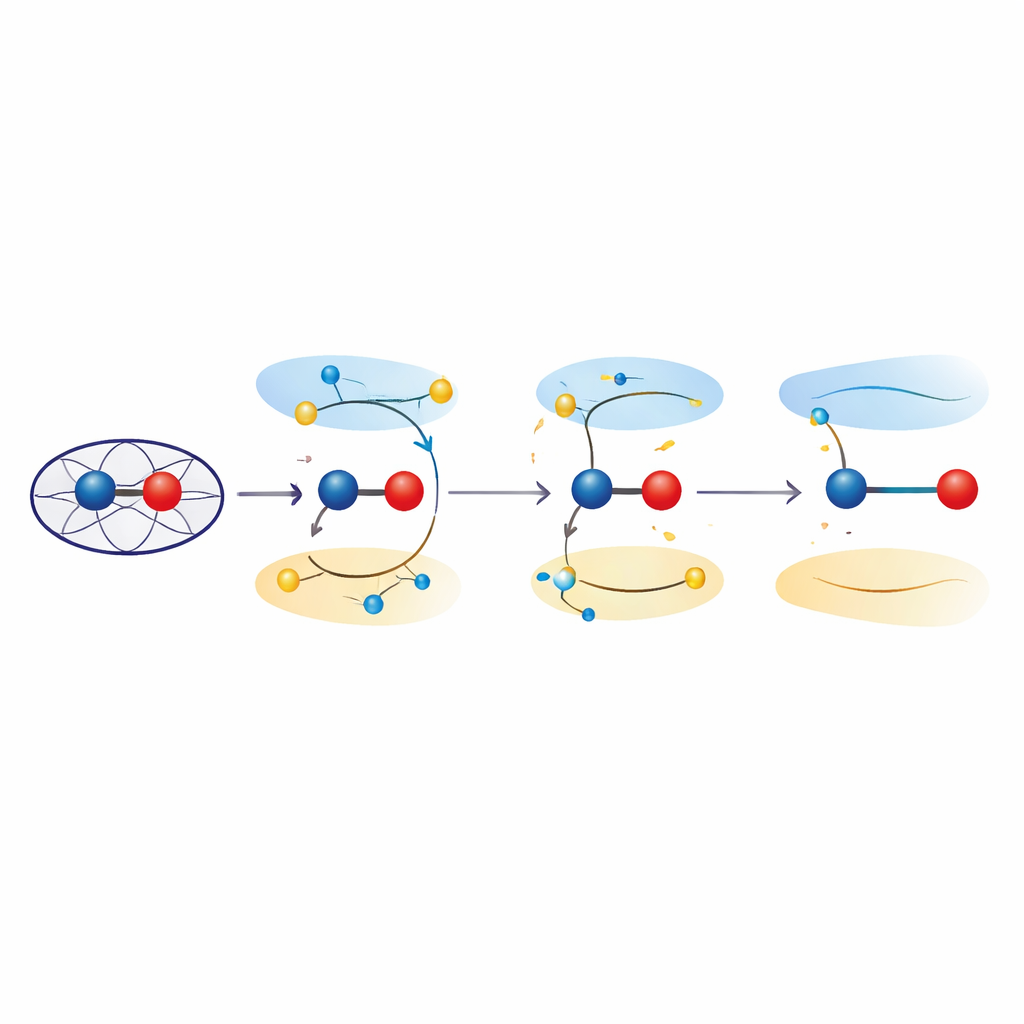
Des états excités qui allongent les liaisons
Pour sonder ces effets, les chercheurs ont eu recours à des calculs de mécanique quantique sur des modèles simplifiés et sur l’environnement protéique complet. En utilisant la théorie de la fonctionnelle de la densité dépendante du temps et des approches combinées mécanique quantique/mécanique moléculaire, ils ont examiné comment la promotion d’électrons d’orbitaux liants vers des orbitales anti‑liantes dans l’unité fer‑oxygène modifie la longueur de liaison préférée. Ces états excités, proches en énergie de l’état fondamental ferryl, ont systématiquement conduit à des distances fer‑oxygène de l’ordre de 1,8–1,9 angström — correspondant aux observations cristallographiques. L’analyse de la distribution électronique a montré que dans ces états, la paire fer‑oxygène ne se comporte plus comme une pure double liaison Fe(IV)=O, mais adopte plutôt un caractère « ferrique‑oxyle », avec des propriétés semblables à un Fe(III) lié à un radical centré sur l’oxygène. Un affinage quantique des structures expérimentales a confirmé que une description en termes d’états excités rend les données au moins aussi bien que les modèles conventionnels en état fondamental.
Ce que cela implique pour la compréhension du pouvoir des enzymes
En termes simples, ce travail suggère que les liaisons fer‑oxygène longues observées dans ces peroxydases hémiques n’exigent pas d’invoquer des dommages, une réduction ou des protons cachés. Elles peuvent plutôt émerger naturellement lorsque l’unité ferryl accède brièvement à des états excités peu élevés qui affaiblissent la liaison et confèrent un caractère ferrique‑oxyle. Pour les non‑spécialistes, cela signifie que la « partie active » de nombreuses enzymes activant l’oxygène pourrait être plus dynamique et électroniquement flexible qu’on ne le pensait, avec de subtils déplacements d’électrons modifiant la force de la liaison et la réactivité sans altérer la chimie globale. Reconnaître ces états excités pourrait remodeler la façon dont les scientifiques interprètent les données structurales sur des oxydants biologiques puissants et guider la conception de catalyseurs artificiels qui imitent, ou ajustent délibérément, cet équilibre électronique délicat.
Citation: Williams, L.J., Kamps, J.J., Brânzanic, A.M.V. et al. Can ferric-oxyl excited states explain elongated iron-oxygen bonds in heme peroxidase catalytic intermediates?. Nat Commun 17, 2324 (2026). https://doi.org/10.1038/s41467-026-69192-8
Mots-clés: peroxydase héminique, intermédiaire ferryl, liaison fer‑oxygène, états électroniques excités, laser à électrons libres X