Clear Sky Science · fr
Un transformeur préentraîné équivariant pour un apprentissage unifié de la représentation moléculaire 3D
Apprendre aux ordinateurs à voir les molécules en 3D
La conception de nouveaux médicaments et matériaux dépend de la compréhension de l’apparence réelle et du mouvement des molécules en trois dimensions, pas seulement de formules planes sur papier. Cet article présente un nouveau modèle d’intelligence artificielle puissant capable d’apprendre à partir des formes 3D de nombreux types de molécules simultanément — des petits composés de type médicament aux grandes protéines et leurs complexes — puis d’utiliser ce savoir pour prédire la force de leurs interactions et lesquels pourraient devenir de futurs médicaments.
Une seule carte pour de nombreux univers moléculaires
La plupart des outils d’IA actuels en chimie sont des spécialistes : l’un est entraîné uniquement sur de petites molécules, un autre seulement sur les protéines, et un troisième uniquement sur leurs complexes. Cette séparation gaspille des données et rend difficile le transfert des connaissances d’un domaine à l’autre. Les auteurs construisent au contraire un seul modèle « fondation », appelé Transformeur Préentraîné Équivariant (EPT), qui apprend à partir d’une vaste collection de structures moléculaires 3D issues de plusieurs bases de données publiques. En traitant toutes ces structures dans un cadre partagé, le modèle peut reconnaître des motifs communs dans l’agencement et les interactions atomiques, qu’il s’agisse d’une simple molécule médicamenteuse ou d’un enchevêtrement complexe de chaînes protéiques. 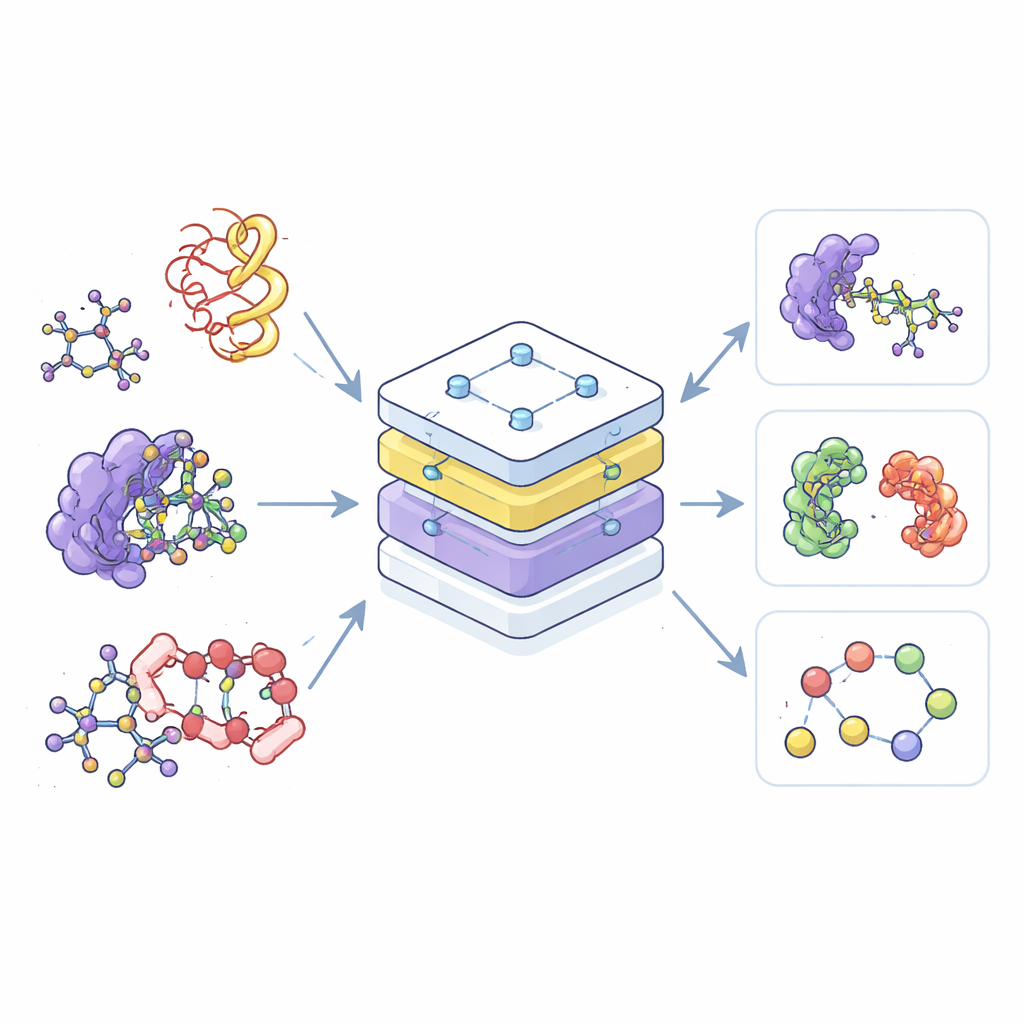
Découper les molécules en morceaux gérables
Pour faire face à la grande variété et à la taille des systèmes moléculaires, les chercheurs introduisent l’idée de « blocs » — de petits morceaux d’atomes porteurs de sens. Pour les petites molécules, un bloc regroupe un atome lourd avec ses hydrogènes attachés ; pour les protéines, chaque acide aminé devient un bloc. Pendant l’entraînement, le modèle voit à la fois les atomes fins et la structure plus grossière en blocs, ce qui lui permet de relier des détails chimiques locaux à des formes 3D plus larges, telles que l’épine dorsale des protéines ou les poches de liaison. Cette vue par blocs crée aussi un langage commun fonctionnant à travers des types moléculaires très différents, rendant possible la compréhension de tous par un seul modèle.
Apprendre en réparant des structures bruitées
Plutôt que d’être guidé par des étiquettes explicites comme « cette molécule est soluble » ou « celle-ci se lie fortement », EPT est entraîné de manière auto-supervisée. Les auteurs perturbent délibérément chaque bloc moléculaire, le déplaçant et le faisant tourner aléatoirement par rapport à sa position réelle, puis demandent au modèle d’inférer les forces et rotations nécessaires pour restaurer la structure originale. Parce que l’entraînement respecte des règles géométriques fondamentales — la molécule doit paraître identique si l’ensemble du système est déplacé ou tourné dans l’espace — le modèle acquiert une notion physiquement cohérente de la forme 3D. Ce jeu de débruitage apprend à EPT comment les atomes se maintiennent au sein et entre les blocs et comment de subtiles variations de géométrie affectent la stabilité. 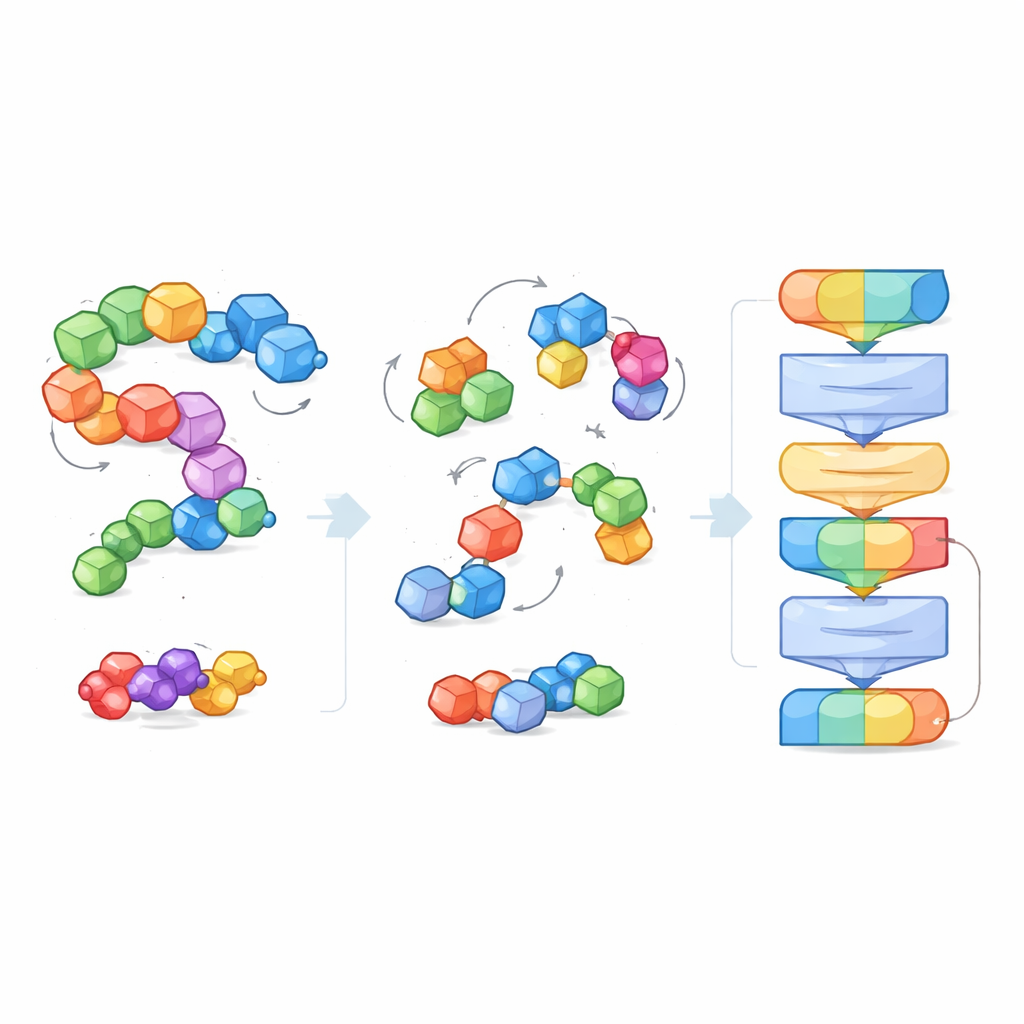
Mettre le modèle à l’épreuve
Après un préentraînement sur plus de cinq millions de structures, EPT est affiné pour plusieurs tâches scientifiques réelles. Il prédit la force de liaison d’une petite molécule à une poche protéique, comment une seule mutation à une interface protéique modifie la liaison, et des propriétés physiques clés de petites molécules qui intéressent les chimistes. Sur des bancs d’essai divers, le modèle unifié égalise ou dépasse les meilleurs outils existants, y compris ceux spécialement adaptés à un seul domaine. Fait notable, entraîné sur un type de données, comme les petites molécules, il aide toujours pour des tâches apparemment différentes — comme la liaison protéique — révélant qu’il a saisi des principes chimiques largement utiles plutôt que des astuces étroites.
À la recherche de nouveaux traitements contre la COVID‑19
Les auteurs démontrent en outre la valeur pratique d’EPT en l’appliquant à un défi de repositionnement de médicaments. Ils affinent d’abord le modèle sur des complexes protéine–ligand, puis l’utilisent pour classer près de 2 000 médicaments déjà approuvés selon leur capacité prédite à se lier à la protéase principale du SARS‑CoV‑2, une enzyme clé dont le virus a besoin pour se répliquer. Des médicaments connus contre la COVID‑19 remontent en haut du classement, et le modèle met en évidence d’autres candidats prometteurs. Douze molécules parmi les mieux classées sont examinées plus en détail par des simulations informatiques, et deux — dont une n’ayant pas été initialement développée pour la COVID‑19 — montrent une liaison prédite particulièrement forte et sont confirmées expérimentalement comme inhibitrices de la protéase virale à des niveaux micromolaires.
Un pas vers une IA moléculaire générale
En termes simples, ce travail montre qu’un seul modèle d’IA conscient de la géométrie peut apprendre une compréhension 3D partagée de nombreux systèmes moléculaires, puis l’utiliser pour répondre à une large gamme de questions scientifiques. En organisant les molécules en blocs et en entraînant le modèle à « réparer » des structures déformées, les auteurs créent un outil qui non seulement prédit des valeurs avec plus de précision, mais peut aussi accélérer des tâches comme la découverte de nouveaux antiviraux. EPT ouvre la voie à un futur où des systèmes d’IA moléculaire polyvalents aideront chimistes et biologistes à explorer l’espace chimique plus efficacement, guidant les expériences et raccourcissant le chemin de la structure atomique aux thérapies et matériaux pratiques.
Citation: Jiao, R., Kong, X., Zhang, L. et al. An equivariant pretrained transformer for unified 3D molecular representation learning. Nat Commun 17, 2606 (2026). https://doi.org/10.1038/s41467-026-69185-7
Mots-clés: représentation moléculaire 3D, transformeur équivariant, découverte de médicaments, liaison protéine–ligand, apprentissage auto-supervisé