Clear Sky Science · fr
Ensembles à résolution atomique de protéines intrinsèquement désordonnées avec AlphaFold
Pourquoi les protéines changeantes comptent
Nos cellules regorgent de protéines qui ne se figent jamais dans une forme unique et rigide. Ces protéines « intrinsèquement désordonnées » se comportent davantage comme des nouilles souples que comme des machines parfaitement repliées, et pourtant elles jouent un rôle central dans des processus allant de la signalisation cellulaire aux maladies neurodégénératives. Parce qu’elles bougent et fléchissent en permanence, capturer l’ensemble de leurs conformations avec un détail atomique est extrêmement difficile et nécessite généralement des années d’expériences et de gros calculs. Cet article présente une nouvelle manière d’exploiter ensemble l’intelligence artificielle et la physique pour cartographier ces molécules agitées beaucoup plus efficacement.
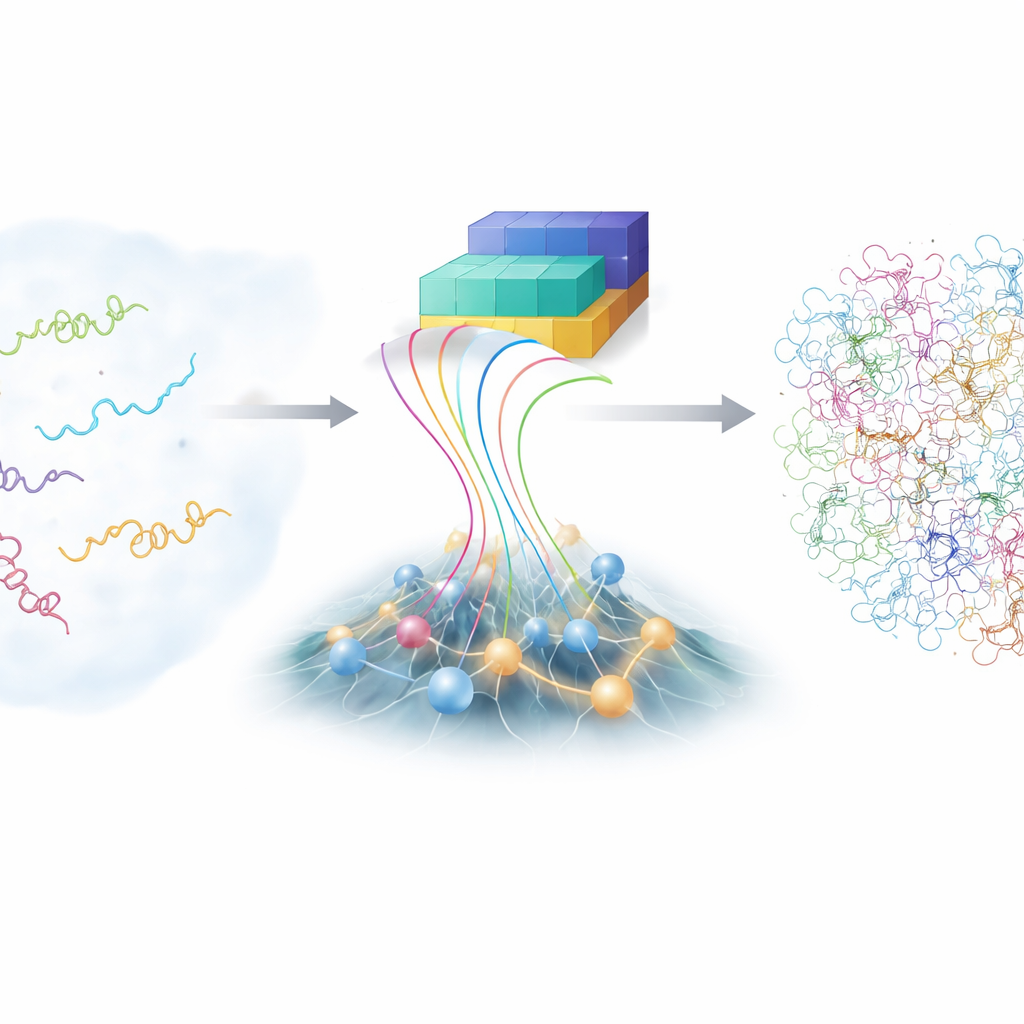
Le défi des molécules agitées
Contrairement aux modèles protéiques des manuels qui montrent une structure ordonnée, les protéines intrinsèquement désordonnées (PID) errent dans un vaste paysage de formes possibles. Cette flexibilité leur permet de reconnaître de nombreux partenaires différents, mais elle les rend aussi notoirement difficiles à étudier. Les techniques de laboratoire traditionnelles, comme la spectroscopie par résonance magnétique nucléaire avancée et la diffusion des rayons X, fournissent des moyennes sur de nombreuses conformations mais pas chaque forme individuelle. Les simulations informatiques à l’échelle atomique peuvent, en principe, suivre chaque atome tandis qu’une PID se tortille, mais elles sont extrêmement coûteuses et dépendent de modèles physiques finement ajustés. En conséquence, la communauté scientifique ne dispose que d’un ensemble limité d’ensembles PID précis et détaillés pour apprendre.
Combiner des estimations intelligentes avec des règles physiques
Ces dernières années, la famille d’outils d’apprentissage profond AlphaFold a stupéfié la biologie en prédisant des structures protéiques à partir de leurs séquences d’acides aminés. Pour les protéines désordonnées, cependant, la force habituelle d’AlphaFold — deviner une seule meilleure forme — est moins utile, car les PID n’ont pas une unique conformation. Ce qu’AlphaFold fournit toutefois, c’est une information riche sur la probabilité que différentes parties de la chaîne soient proches ou éloignées les unes des autres. Les auteurs ont construit un nouveau cadre, appelé bAIes, qui traite ces informations issues de l’IA comme une guidance souple et les mélange à un modèle physique rapide qui part délibérément d’une vue de « rouleau aléatoire », où la chaîne explore tous les plis et torsions possibles sans favoriser de structure particulière.
Des enchevêtrements aléatoires à des ensembles réalistes
Premièrement, les chercheurs ont construit un modèle physique efficace qui reproduit le comportement d’une chaîne protéique complètement non structurée, à partir de statistiques extraites de milliers de structures protéiques connues. Ce modèle sert de « prior » — l’attente de base sur la façon dont une PID se déplace si l’on ne sait rien d’autre. Ensuite, bAIes lit les prédictions d’AlphaFold sur les paires de résidus susceptibles de se rapprocher. Plutôt que de forcer la protéine dans un motif unique, il convertit ces indices en contraintes de distance douces avec une incertitude intégrée, permettant à la chaîne de satisfaire les suggestions de l’IA seulement lorsqu’elles sont compatibles avec le cadre physique plus large.
Testé contre des expériences réelles
Pour vérifier l’efficacité de cette approche, l’équipe a appliqué bAIes à un ensemble de 21 protéines allant de chaînes presque entièrement aléatoires à des systèmes plus complexes avec des hélices transitoires et plusieurs domaines. Pour chacune, ils ont comparé les ensembles générés par ordinateur à une large série de mesures expérimentales qui sondent à la fois les détails locaux et la taille et la forme globales. Pour des protéines très souples comme le peptide lié à la maladie d’Alzheimer Aβ40, le modèle simple de rouleau aléatoire était déjà proche de la réalité, et bAIes a préservé ce bon accord. Pour les protéines partiellement structurées, bAIes a amélioré la concordance avec les expériences en capturant correctement où de courts segments hélicoïdaux et des zones compactes apparaissent et disparaissent. Fait crucial, la méthode est restée robuste même lorsqu’AlphaFold était trop confiant et prédisait à tort des reploiements stables alors que les expériences en solution montrent du désordre, parce que bAIes permet explicitement des erreurs dans l’entrée de l’IA.
Surpasser ou égaler les méthodes existantes
Les auteurs ont ensuite opposé bAIes à des simulations tout-atomes lourdes exécutées sur des superordinateurs spécialisés, à des modèles grossiers qui simplifient les protéines en perles, et à de nouveaux générateurs d’apprentissage profond entraînés sur des données de simulation. Lors de multiples tests, bAIes a systématiquement égalé ou surpassé ces approches pour reproduire les données expérimentales, tout en étant beaucoup moins exigeant en ressources informatiques que les simulations à grande échelle. Il a également fonctionné au-delà des PID simples, traitant des protéines avec plusieurs domaines rigides reliés par des connecteurs flexibles et retrouvant leur forme globale en solution. Lorsque les chercheurs ont affiné davantage les ensembles bAIes avec des données expérimentales, l’accord s’est encore amélioré, montrant que la méthode peut servir de point de départ puissant pour la modélisation intégrative.
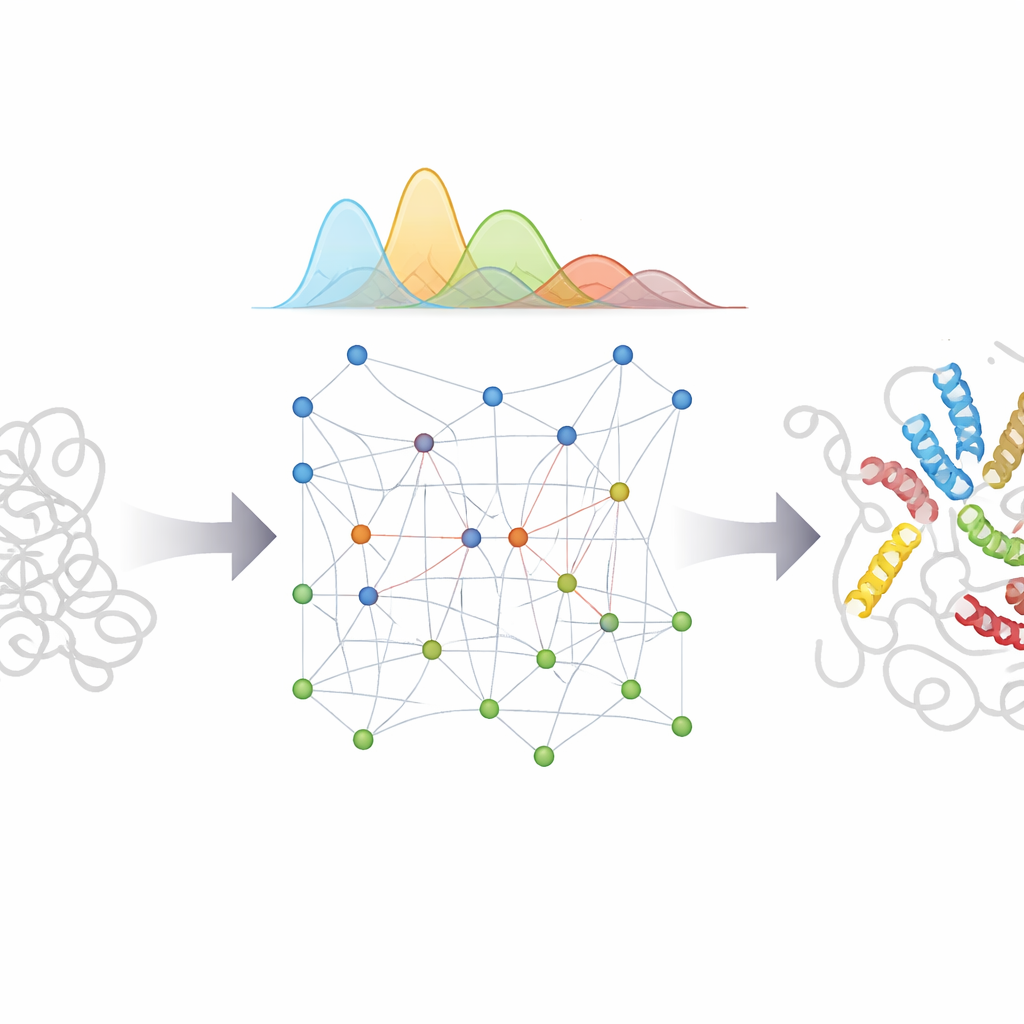
Ce que cela signifie pour la biologie et la médecine
En mariant la puissance de reconnaissance de motifs d’AlphaFold avec un modèle physique soigneusement conçu et un traitement bayésien de l’incertitude, bAIes offre une voie pratique vers des « films » détaillés de protéines désordonnées plutôt que des instantanés uniques. Ces ensembles détaillés au niveau atomique peuvent aider les scientifiques à comprendre comment des régions flexibles reconnaissent des partenaires, comment le mauvais repliement et l’agrégation commencent dans des maladies comme Parkinson et Alzheimer, et comment de petites molécules pourraient se lier à des cibles insaisissables et changeantes. Parce que la méthode est efficace et intégrée dans un logiciel open source, elle peut être largement adoptée pour générer des ensembles réalistes pour de nombreuses protéines désordonnées, orienter les expériences et soutenir de futurs systèmes d’IA qui visent à prédire non pas une seule structure, mais l’éventail complet de formes que peuvent adopter les molécules les plus flexibles de la vie.
Citation: Schnapka, V., Morozova, T.I., Sen, S. et al. Atomic resolution ensembles of intrinsically disordered proteins with Alphafold. Nat Commun 17, 2399 (2026). https://doi.org/10.1038/s41467-026-69172-y
Mots-clés: protéines intrinsèquement désordonnées, AlphaFold, modélisation bayésienne, ensembles protéiques, biologie structurale