Clear Sky Science · fr
scLong : un modèle fondamental d’un milliard de paramètres pour capturer le contexte génique à longue portée en transcriptomique unicellulaire
Apprendre aux ordinateurs à lire le langage caché des cellules
Chaque cellule de votre corps abrite une véritable cité de gènes qui s’activent et se désactivent selon des motifs complexes. Le séquençage ARN unicellulaire moderne peut maintenant écouter chaque cellule individuellement, mais le résultat est un flot écrasant de chiffres. Cet article présente scLong, un modèle d’intelligence artificielle massif conçu pour donner du sens à ces motifs d’activité génique complexes, y compris aux signaux faibles que les méthodes antérieures ont tendance à ignorer. Son objectif est d’aider les chercheurs à comprendre comment les cellules réagissent lorsque des gènes sont modifiés, des médicaments ajoutés ou des maladies s’installent.
Pourquoi les cartes géniques au niveau cellulaire sont importantes
Les études génétiques traditionnelles mélangent souvent des millions de cellules, moyennant les différences et effaçant les états rares ou atypiques. Les techniques unicellulaires ont changé cela en mesurant l’activité des gènes pour chaque cellule individuellement, révélant des types cellulaires cachés, des communications subtiles entre cellules et des circuits de contrôle détaillés qui déterminent le devenir d’une cellule. Cependant, analyser ce type de données est extrêmement difficile : chaque cellule peut comporter des niveaux d’activité pour des dizaines de milliers de gènes, dont beaucoup sont à peine détectables. Les modèles d’IA existants simplifient le problème en ne se concentrant que sur les gènes les plus exprimés, ce qui accélère le calcul mais fait manquer de nombreux signaux subtils qui pourraient être cruciaux en cas de maladie, de développement ou de réponse aux traitements.
Un nouveau modèle qui écoute chaque gène
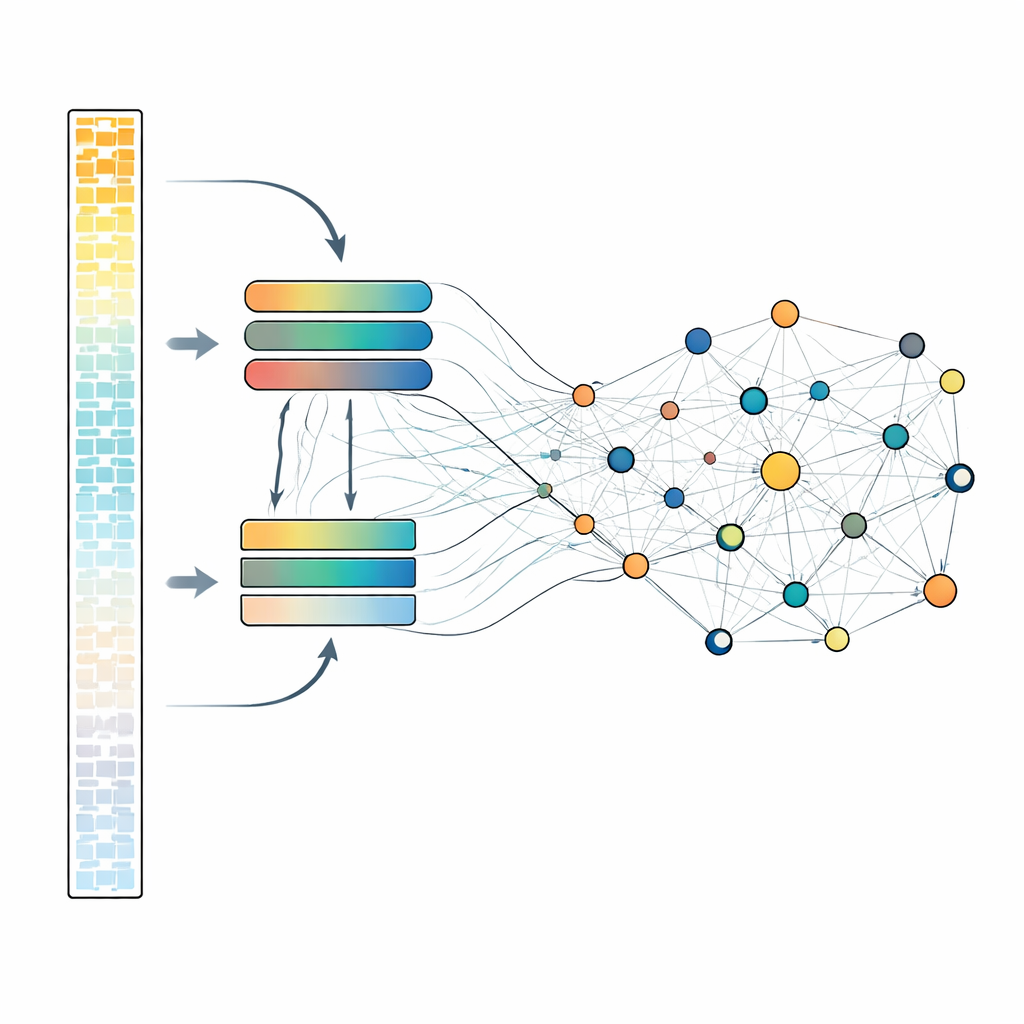
scLong relève ce défi en augmentant l’échelle plutôt qu’en réduisant la donnée. C’est un modèle fondamental d’un milliard de paramètres entraîné sur des profils d’activité génique issus d’environ 48 millions de cellules humaines provenant de plus de 50 tissus. Contrairement aux approches antérieures qui se limitent à quelques milliers de gènes très actifs, scLong considère environ 28 000 gènes à la fois, y compris ceux qui sont rarement ou faiblement exprimés. Il combine deux types d’informations pour chaque gène : son niveau d’activité dans une cellule donnée et ce que l’on sait déjà de sa fonction via l’Ontology des gènes (Gene Ontology), un vaste catalogue expert et organisé des rôles et relations des gènes. Un réseau spécialisé opérant sur un graphe de connexions géniques distille ces connaissances a priori en représentations compactes que le modèle peut utiliser en parallèle des valeurs d’expression brutes.
Comment le modèle équilibre puissance et efficacité
Examiner chaque gène en détail est coûteux en calcul, donc scLong adopte une conception astucieuse à deux voies. À l’intérieur de chaque cellule, les gènes sont triés selon l’intensité de leur expression. Les gènes les plus actifs, qui portent souvent le signal biologique principal, sont traités par un module d’attention plus grand et plus puissant. Les gènes plus silencieux, y compris ceux avec des mesures faibles voire nulles, sont dirigés vers un module plus petit et plus léger. Ensuite, tous les gènes sont recombinés et passés par une autre couche d’attention qui permet à chaque gène d’influencer tous les autres. Cette architecture permet au modèle de conserver des représentations moins coûteuses mais néanmoins informatives pour les signaux faibles tout en réservant plus de capacité pour les signaux forts. Lors du préentraînement, le système masque de façon répétée un sous-ensemble de valeurs d’expression génique et apprend à les reconstruire à partir du contexte environnant, l’obligeant à découvrir les motifs qui relient les gènes entre eux.
Mettre le modèle au travail sur des problèmes réels
Une fois entraîné, scLong peut être adapté à un large éventail de questions biologiques. Les auteurs montrent qu’il prédit comment l’activité génique changera lorsque des gènes spécifiques sont désactivés ou modifiés, y compris des combinaisons de deux gènes susceptibles d’agir de concert. Il prévoit également comment les cellules réagissent lorsqu’elles sont exposées à différents composés chimiques, ce qui est important pour la découverte et l’évaluation de médicaments. Dans des études sur le cancer, scLong aide à anticiper la réponse des lignées tumorales à des médicaments simples et à des paires de médicaments qui pourraient mieux fonctionner en combinaison, surpassant souvent à la fois des modèles spécialisés et d’autres grands modèles fondamentaux. Au-delà de la prédiction, scLong peut inférer des réseaux de relations régulatrices entre gènes et aider à corriger les distorsions techniques qui surviennent lorsque les données sont collectées dans différents laboratoires ou sur des machines différentes.
Ce que cela signifie pour la médecine et la recherche futures
En termes simples, scLong offre aux scientifiques une carte de l’activité génique à haute résolution et sensible au contexte au sein des cellules individuelles, sans éliminer les gènes peu expressés ou rarement utilisés. En apprenant à partir de millions de cellules et en incorporant les connaissances biologiques existantes, il propose des estimations plus précises de la façon dont les cellules réagiront lorsque des gènes seront perturbés, lorsque de nouveaux médicaments seront introduits ou lorsque des processus pathologiques se développeront. Cela pourrait accélérer la recherche de nouvelles thérapies, orienter des choix de traitement plus personnalisés et affiner notre compréhension de la façon dont des réseaux géniques complexes contrôlent la santé et la maladie. Bien que le modèle soit vaste et exigeant en ressources de calcul, il ouvre la voie à un futur où des systèmes d’IA puissants et polyvalents servent de compagnons pour explorer le fonctionnement caché de nos cellules.
Citation: Bai, D., Mo, S., Zhang, R. et al. scLong: a billion-parameter foundation model for capturing long-range gene context in single-cell transcriptomics. Nat Commun 17, 2380 (2026). https://doi.org/10.1038/s41467-026-69102-y
Mots-clés: transcriptomique unicellulaire, modèles fondamentaux, régulation des gènes, prédiction de la réponse aux médicaments, expression génique