Clear Sky Science · fr
Prédiction de similarité chimique entre spectres de masse en mode d’ionisation croisée en métabolomique
Pourquoi il est important de relier les points chimiques
Chaque gorgée de café, bouffée d’air ou prise de médicament laisse de minuscules traces chimiques dans nos organismes. Les instruments modernes peuvent détecter des milliers de ces molécules simultanément, mais transformer ces signaux en informations biologiques reste étonnamment difficile. Cette étude présente MS2DeepScore 2.0, un outil d’apprentissage automatique qui aide les scientifiques à voir comment ces molécules sont liées, même lorsque les signaux sont enregistrés de façons très différentes. Ce faisant, il promet des interprétations plus rapides et plus complètes des mélanges chimiques complexes en médecine, nutrition et recherche environnementale.
Deux façons d’observer la même molécule
La spectrométrie de masse est une technique essentielle qui pèse et fragmente les molécules pour révéler leur identité. Dans les expériences courantes, les chercheurs mesurent souvent le même échantillon en deux modes : l’un favorisant les molécules chargées positivement et l’autre les molécules chargées négativement. Chaque mode produit sa propre « empreinte » caractéristique de fragments. Même lorsque les deux mesures proviennent de la même molécule, les motifs résultants peuvent paraître si différents que les méthodes de comparaison traditionnelles échouent. En conséquence, les chercheurs analysent généralement les deux modes séparément, construisent deux cartes déconnectées de l’échantillon et risquent de manquer des relations chimiques importantes.
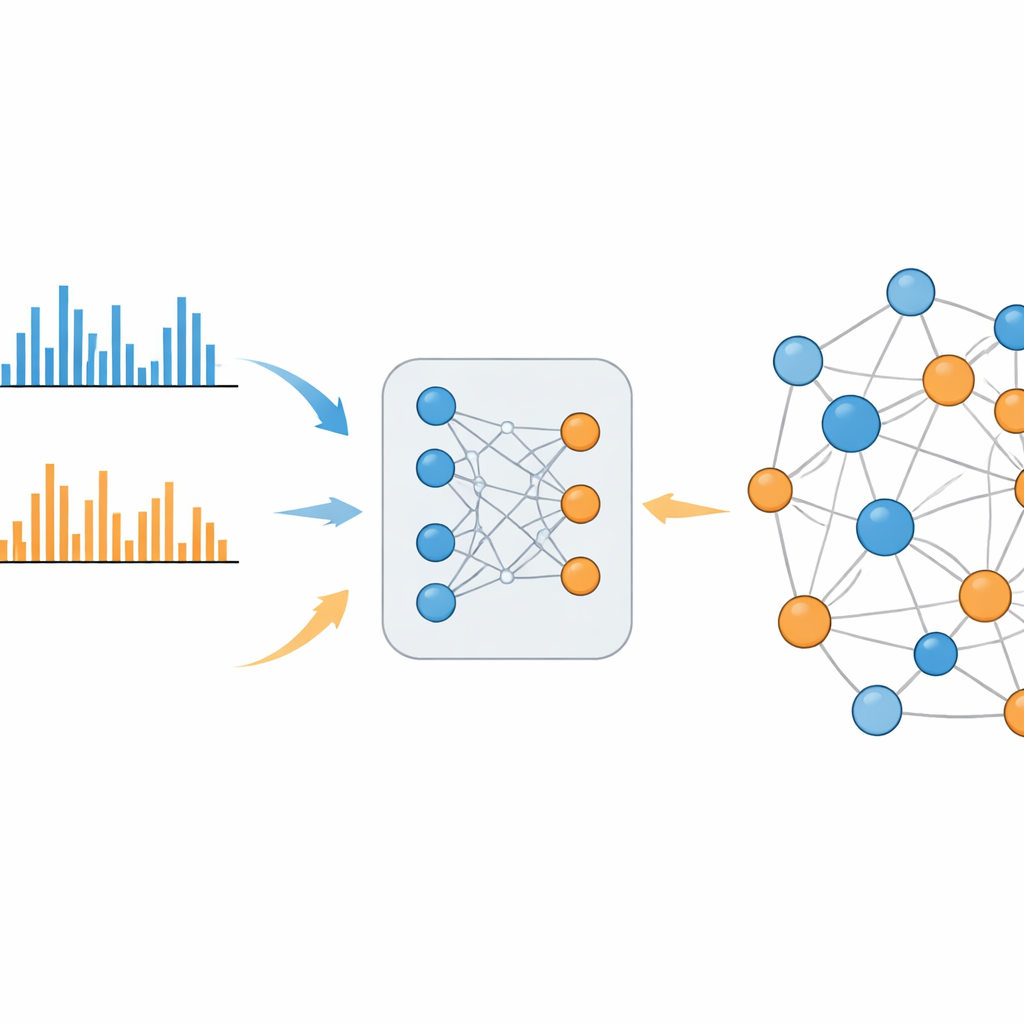
Un système d’apprentissage qui comble le fossé
MS2DeepScore 2.0 s’attaque à cette division en apprenant la similarité chimique directement à partir de larges bibliothèques de spectres connus. Le modèle repose sur une architecture de réseaux neuronaux jumeaux qui convertit chaque motif de fragmentation en une empreinte de 500 nombres, appelée embedding. Pendant l’entraînement, le système voit des centaines de milliers d’exemples issus des modes positif et négatif, ainsi que la similarité réelle des molécules sous-jacentes. Il s’ajuste pour que les spectres de molécules apparentées aboutissent à des embeddings proches, qu’ils aient été mesurés dans le même mode ou dans des modes opposés. La nouvelle version dépasse son prédécesseur en intégrant des informations supplémentaires, telles que la masse de la molécule d’origine et le mode d’ionisation utilisé, et en appliquant un schéma d’échantillonnage soigneusement équilibré afin que des relations chimiques rares mais informatives ne soient pas noyées par des relations courantes et peu pertinentes.
Des signaux dispersés à des cartes unifiées
Une fois entraîné, MS2DeepScore 2.0 peut estimer la similarité chimique de n’importe quelles deux spectra, y compris des paires mode positif versus négatif. Les auteurs montrent que ces prédictions corrèlent bien avec des mesures établies de similarité structurale, non seulement au sein de chaque mode, mais aussi entre modes. En utilisant des données réelles d’urine humaine, de plasma sanguin humain et d’une plante comestible sauvage, ils construisent des « réseaux moléculaires » où chaque spectre est un nœud et une forte similarité prédite crée une connexion. Contrairement aux approches plus anciennes, ces réseaux mélangent naturellement les données des modes positif et négatif en cartes uniques et cohérentes. Des grappes annotées par des experts incluent, par exemple, des groupes de molécules liées à la caféine dans l’urine qui sont connectés à travers les modes d’ionisation et correspondent à des voies métaboliques connues.
Voir le paysage chimique d’un coup d’œil
Les réseaux moléculaires sont puissants mais peuvent devenir embrouillés si trop de liens faibles sont inclus. Pour éviter cela, les auteurs utilisent les embeddings de MS2DeepScore directement comme coordonnées dans une mise en page bidimensionnelle créée avec une technique appelée UMAP. Chaque point de cette carte représente un spectre, et les points voisins correspondent à des molécules que le modèle considère comme chimiquement similaires. Les spectres des modes positif et négatif d’un même composé, qui paraissent très différents à l’œil nu, se retrouvent souvent côte à côte dans cet espace d’embedding. L’équipe entraîne également un modèle additionnel qui inspecte chaque embedding et évalue sa fiabilité, signalant les spectres bruyants, incomplets ou différents de tout ce qui a été vu lors de l’entraînement. Éliminer ces points de faible qualité améliore la précision globale et rend les visualisations plus fiables.
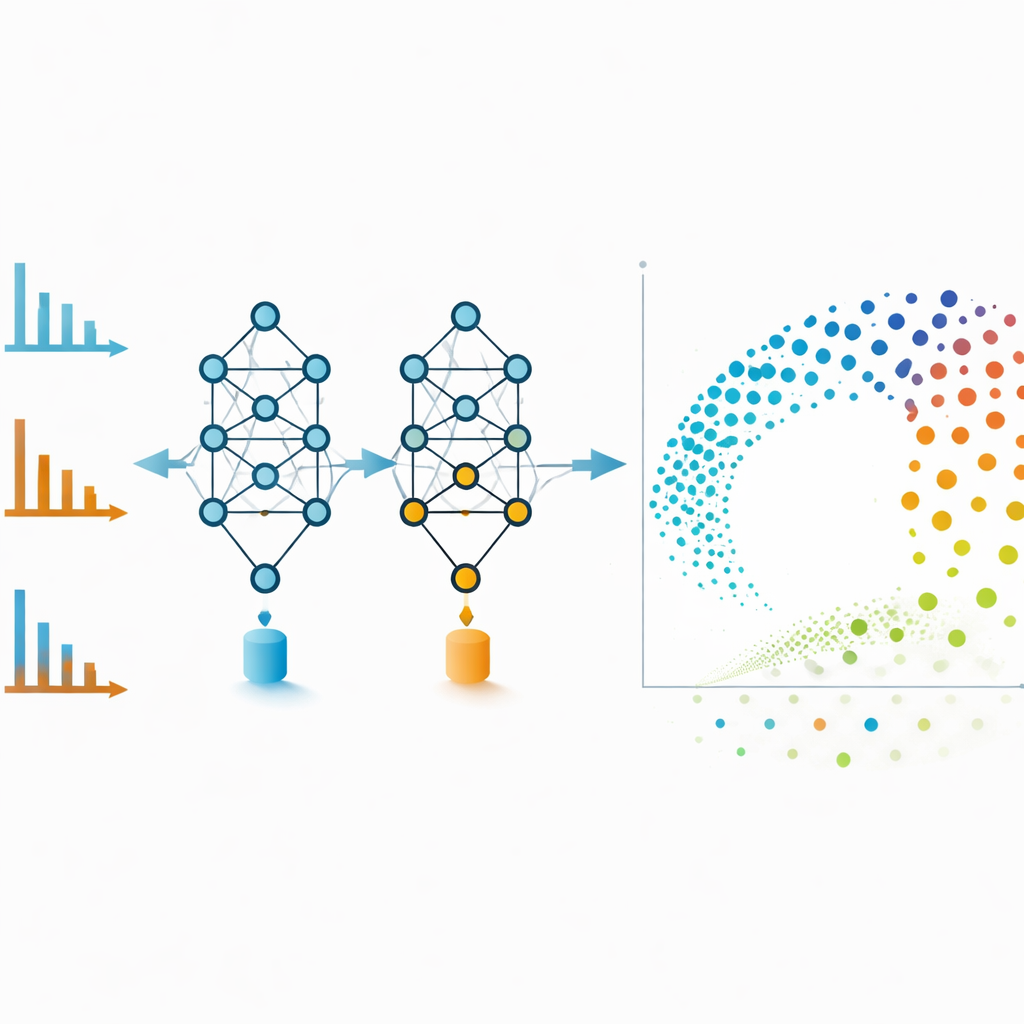
Apporter des outils avancés aux laboratoires quotidiens
Pour garantir que cette technologie soit utilisable au-delà des experts en programmation, les auteurs ont intégré MS2DeepScore 2.0 dans des logiciels de spectrométrie de masse populaires et gratuits. Grâce à cette intégration, les chercheurs peuvent détecter des caractéristiques, construire des réseaux moléculaires qui ignorent les frontières entre modes d’ionisation et explorer l’espace chimique résultant via des tableaux de bord interactifs. Le code, les modèles entraînés et les jeux de données d’exemple sont partagés ouvertement, et le système peut être réentraîné ou affiné pour des classes chimiques spécialisées.
Qu’est-ce que cela signifie pour les découvertes à venir
Pour les non-spécialistes, le message clé est que MS2DeepScore 2.0 aide à transformer des mesures fragmentées et dépendantes du mode en une image unique et plus compréhensible des molécules présentes dans un échantillon. En reliant de manière fiable des signaux qui vivaient auparavant dans des mondes analytiques séparés, la méthode permet aux scientifiques d’exploiter des bibliothèques de référence beaucoup plus larges, de comparer les échantillons de façon plus complète et de concentrer leur attention sur des grappes pertinentes de composés apparentés. Cette interconnexion des données devrait accélérer l’identification de biomarqueurs, nutriments, produits naturels et polluants, approfondissant en fin de compte notre compréhension de la manière dont la chimie influence la santé et l’environnement.
Citation: de Jonge, N.F., Chekmeneva, E., Schmid, R. et al. Cross ionization mode chemical similarity prediction between tandem mass spectra in metabolomics. Nat Commun 17, 2483 (2026). https://doi.org/10.1038/s41467-026-69083-y
Mots-clés: métabolomique, spectrométrie de masse, apprentissage automatique, mise en réseau moléculaire, similarité chimique