Clear Sky Science · fr
FANCD2 freine la progression des fourches et prévient la fragilité aux origines précoces lors de la re-réplication
Quand la copie de l’ADN dévie légèrement du script
À chaque division, une cellule doit copier exactement une fois l’intégralité de sa bibliothèque d’ADN. Si des portions de cette bibliothèque sont copiées deux fois, ou copiées de manière précipitée et approximative, le résultat peut être des chromosomes fracturés et des mutations qui alimentent le cancer. Cette étude examine ce qui se passe lorsque les garde-fous cellulaires contre des cycles de copie supplémentaires commencent à céder, et révèle comment une protéine de réparation, FANCD2, intervient pour empêcher des cellules légèrement défaillantes de basculer dans un chaos génomique majeur.
Garde-corps pour une seule copie propre
Nos chromosomes sont dupliqués à partir de milliers de points de départ, ou « origines », qui sont autorisés puis activés dans une séquence minutieusement synchronisée. Une petite protéine nommée Geminin aide normalement à garantir que chaque origine ne s’active qu’une seule fois par cycle cellulaire. Lorsque Geminin est perdue ou affaiblie, certaines origines peuvent se réactiver sur de l’ADN déjà copié, une situation connue sous le nom de re-réplication. Les cellules cancéreuses, qui surexpriment souvent des facteurs d’activation, sont particulièrement susceptibles à ce problème. Les auteurs ont d’abord utilisé un criblage génétique à haut contenu dans des cellules humaines prédisposées à une faible re-réplication par déplétion de Geminin. Ils ont recherché quels gènes de réparation de l’ADN et de contrôle du cycle deviennent cruciaux dans cet état de stress et ont trouvé que FANCD2, surtout connu pour réparer les liaisons croisées de l’ADN dans l’anémie de Fanconi, émerge comme un protecteur clé de la survie cellulaire et de l’intégrité du génome.
Un premier intervenant aux machines de copie surchargées
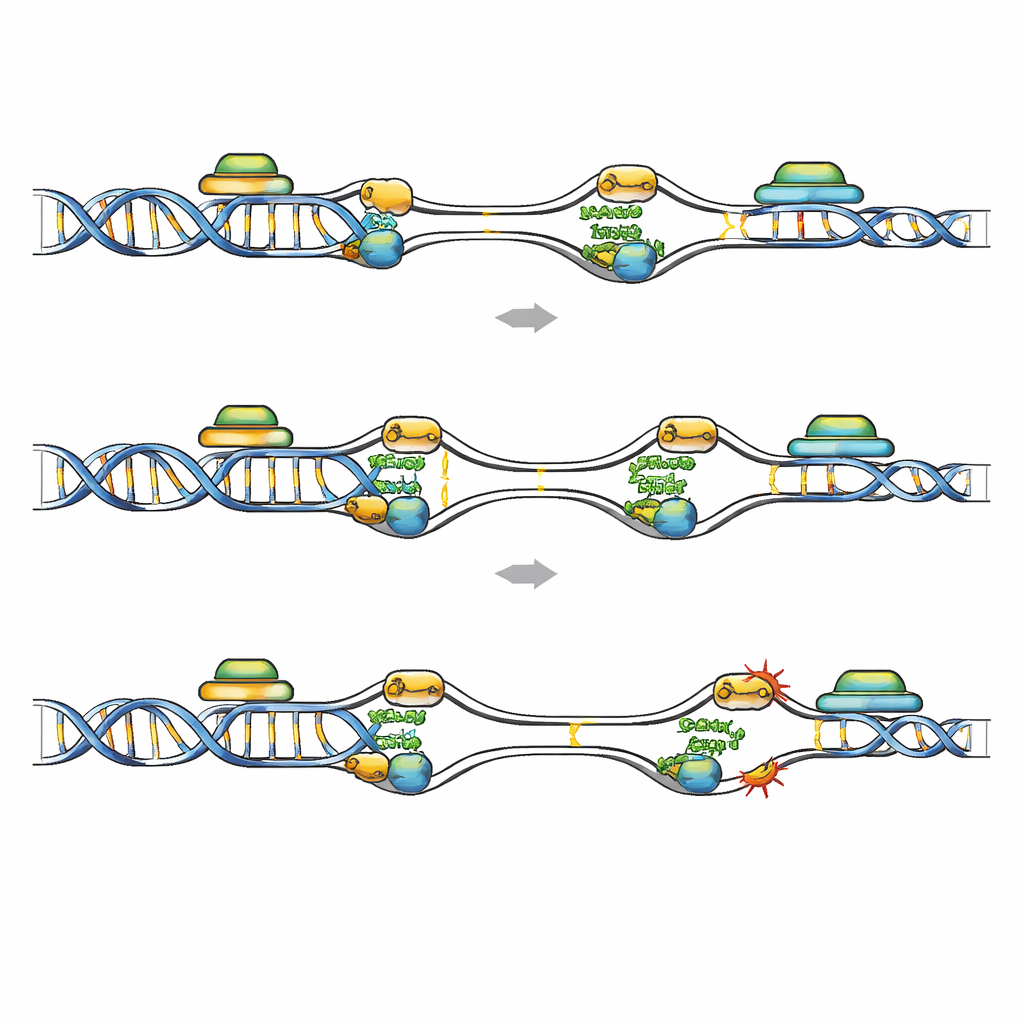
L’équipe a ensuite suivi où et quand FANCD2 apparaît dans des cellules en re-réplication. Peu après la disparition de Geminin, FANCD2 s’accumule rapidement sur la chromatine et forme de vives foyers nucléaires, bien avant que des cassures d’ADN généralisées ne soient détectables. Grâce au marquage de l’ADN nouvellement synthétisé associé à des essais de proximité, ils ont montré que FANCD2 est recruté directement aux fourches de réplication actives, en particulier dans les cellules dont l’ADN est déjà en train d’être copié une seconde fois. Dans des cellules synchronisées relâchées dans le cycle cellulaire suivant, une population distincte présentant un motif diffus d’ADN sur-répliqué est apparue. Ces cellules affichaient des signaux forts de FANCD2 et de RPA, indiquant un stress de réplication en cours, et étaient bloquées à la frontière avant la mitose par un point de contrôle actif, ce qui suggère que FANCD2 fait partie d’une réponse précoce qui stabilise les fourches stressées plutôt que de simplement réagir à l’ADN cassé.
Retenir les fourches débridées et les lacunes cachées
Pour tester comment FANCD2 influe sur la copie de l’ADN, les chercheurs ont combiné la perte de Geminin avec la déplétion de FANCD2. De manière surprenante, supprimer FANCD2 n’a pas augmenté la fraction de cellules présentant des génomes manifestement re-répliqués. En revanche, des essais sur fibres d’ADN monomoléculaires ont révélé que les fourches de réplication parcouraient de plus grandes distances et devenaient plus asymétriques, signe d’une progression inégale et instable. Ces fourches plus rapides laissaient derrière elles davantage de lacunes simple-brin dans l’ADN nouvellement synthétisé, visibles comme des foyers intenses de RPA et de BrdU natif et confirmées par la sensibilité des tronçons marqués à une enzyme qui coupe les régions simple-brin. Les cellules dépourvues à la fois de Geminin et de FANCD2 présentaient une hausse des cassures chromosomiques, des fragments, des corps nucléaires et des micronoyaux, tous signes d’une instabilité génomique sévère. Le blocage de PARP, un facteur qui aide normalement à gérer de telles lacunes, a imité et aggravé ces défauts, soulignant que la formation incontrôlée de lacunes est centrale dans les dommages observés.
Points chauds fragiles où copie et transcription entrent en collision
Le cartographie à l’échelle du génome des sites de liaison de FANCD2 a révélé où la re-réplication est la plus dangereuse. Dans des cellules leucémiques appauvries en Geminin, FANCD2 s’est déplacée des sites fragiles communs classiques vers des origines de réplication à activation précoce, intégrées dans de courts gènes riches en GC et fortement transcrits. Ces régions portent des marques de transcription active et sont sujettes aux R-boucles, où l’ARN naissant s’hybride avec son ADN matrice, pouvant bloquer la réplication. Des jeux de données publics ont montré plus de dommages à l’ADN et des signaux accrus d’hybrides ARNm–ADN dans les gènes enrichis en FANCD2 après perte de Geminin, et ces régions recouvraient les soi‑disant sites fragiles précocement répliqués. Lorsque la transcription a été globalement atténuée par un médicament, ou lorsque les R-boucles ont été spécifiquement éliminées en surexprimant RNase H1, le nombre de foyers de FANCD2, RPA et de dommages à l’ADN dans les cellules déficientes en Geminin a chuté nettement. Cela indique que les collisions entre origines réactivées et unités de transcription actives, amplifiées par les R-boucles, créent des points chauds fragiles que FANCD2 doit protéger.
Affiner la protection par des étiquettes chimiques
FANCD2 est activée en partie par l’ajout d’un petit tag de type ubiquitine. En appauvrissant FANCA, un composant central de la machinerie d’étiquetage, et en utilisant des cellules exprimant un mutant de FANCD2 résistant à cette modification, les auteurs ont montré que la mono‑ubiquitination améliore la survie des cellules en re-réplication mais n’est pas absolument requise. Même une FANCD2 non étiquetée apportait une protection partielle, cohérente avec des rôles distincts à la fois dans la détection et la stabilisation des fourches stressées. Le tableau d’ensemble est que FANCD2 aide à ralentir et organiser la réplication aux origines précoces vulnérables et limite combien et à quelle ampleur les lacunes simple-brin se forment.
Pourquoi cela importe pour le traitement du cancer
Pour les non-spécialistes, le message central est que toutes les erreurs de réplication ne sont pas catastrophiques dès le départ. Une re-réplication légère, comme c’est le cas dans certains tumeurs, peut être tolérée si des systèmes protecteurs comme FANCD2 maintiennent la copie d’ADN sous contrôle et empêchent que des lacunes fragiles ne se transforment en chromosomes brisés. Lorsque ce garde-fou est retiré ou submergé, les mêmes erreurs mineures d’autorisation s’aggravent rapidement en fragmentation du génome. Parce que la perte de Geminin et les défauts d’autorisation de la réplication sont enrichis dans les cellules cancéreuses, et que de nombreuses tumeurs présentent déjà des faiblesses dans le réseau Fanconi/BRCA, les vulnérabilités mises au jour ici suggèrent des stratégies thérapeutiques : combiner des inhibiteurs qui poussent les cellules cancéreuses vers la re-réplication avec des médicaments qui exacerbent l’accumulation de lacunes, comme les inhibiteurs de PARP, pourrait conduire sélectivement les cellules malignes au-delà de leur seuil de tolérance tout en épargnant les cellules normales dotées d’une protection intacte.
Citation: Badra-Fajardo, N., Karydi, E., Bayona-Feliu, A. et al. FANCD2 restrains fork progression and prevents fragility at early origins upon re-replication. Nat Commun 17, 2478 (2026). https://doi.org/10.1038/s41467-026-68966-4
Mots-clés: Stress de la réplication de l’ADN, FANCD2, Geminin, re-réplication, instabilité du génome