Clear Sky Science · fr
Un modèle polymère éclairé par l’expérimentation révèle l’organisation à haute résolution des loci génomiques
Comment le repliement de l’ADN façonne l’identité cellulaire
Toutes les cellules de votre corps portent essentiellement le même ADN, et pourtant les cellules du cerveau, de la peau ou les cellules souches se comportent de façon très différente. Une raison essentielle tient à la manière dont cet ADN est replié et compacté dans le noyau. Cette étude introduit une nouvelle façon de « voir » ce repliement avec un niveau de détail remarquable, en reliant l’agencement physique de l’ADN à l’état d’activation des gènes importants. En combinant données expérimentales et simulations informatiques basées sur la physique, les auteurs dévoilent des amas cachés de matière génétique qui semblent agir comme des briques élémentaires de l’organisation du génome.
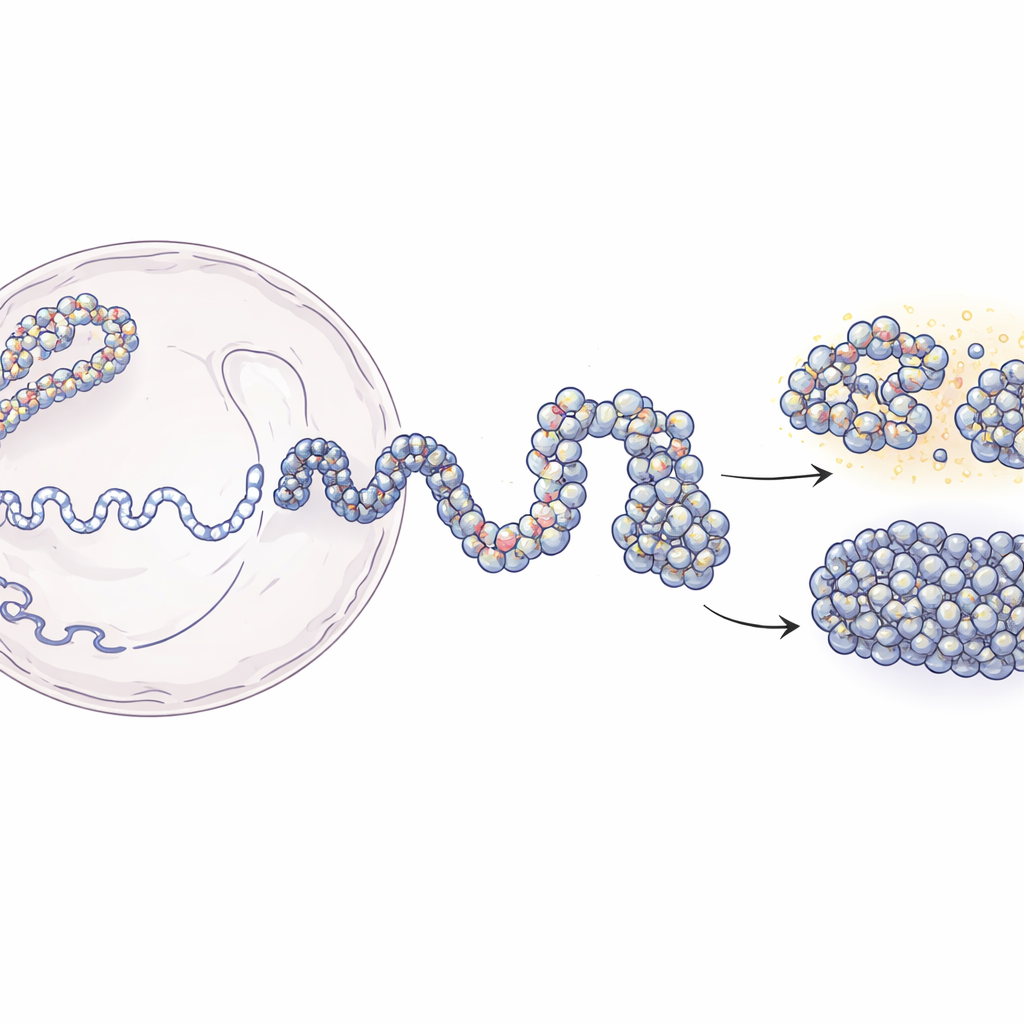
Des longs brins d’ADN aux cartes 3D du génome
Dans le noyau, l’ADN s’enroule autour de protéines en forme de bobines appelées nucléosomes, formant une structure « perles sur un fil » connue sous le nom de chromatine. Les techniques modernes comme Hi-C et Micro-C renseignent sur les fragments d’ADN qui se trouvent proches en 3D, mais fournissent en général des images floues moyennées sur une population de cellules. À l’inverse, les expériences qui localisent les nucléosomes individuellement donnent des détails locaux précis mais peu d’informations sur la structure à grande échelle. Ce travail comble ce fossé. Les auteurs partent de cartes de contacts à basse résolution qui indiquent la fréquence de rencontre entre segments d’ADN distants, puis les combinent avec des cartes expérimentales de positions de nucléosomes. En appliquant des principes de la physique des polymères, ils construisent des ensembles 3D simulés de chromatine qui concordent avec les données expérimentales tout en résolvant des structures jusqu’à quelques dizaines de bases d’ADN.
Une stratégie en deux étapes pour reconstruire la chromatine
L’approche de modélisation se déploie en deux grandes étapes. D’abord, l’équipe utilise des données Hi-C pour générer de nombreuses formes possibles à grande échelle d’une portion d’ADN de 200 000 bases, traitant la chromatine comme une chaîne flexible où des segments de 5 000 bases sont guidés pour établir ou éviter des contacts observés expérimentalement. Ces structures grossières captent le motif de repliement global que les protéines cellulaires contribuent à créer. Dans la seconde phase, chaque grosse « perle » est remplacée par une chaîne beaucoup plus fine composée de nucléosomes individuels et des courts segments d’ADN qui les relient. Les positions de ces nucléosomes proviennent d’une méthode de cartographie basée sur une enzyme (MNase-seq) qui révèle où ils se situent typiquement le long du génome. Les chaînes fines sont alors laissées se replier tout en respectant l’architecture plus large. Quand les chercheurs « floutent » leurs modèles haute résolution jusqu’aux résolutions expérimentales, ils reproduisent avec une grande précision les cartes de contacts Hi-C et Micro-C.
Découverte des « blobs » de nucléosomes comme unités structurelles
Lorsqu’ils ont zoomé sur leurs structures simulées, un motif frappant est apparu : les nucléosomes n’étaient pas répartis de manière homogène, mais rassemblés en agrégats irréguliers que les auteurs appellent des blobs de nucléosomes. Ces blobs ressemblent aux structures en amas observées précédemment dans des images en super-résolution de cellules réelles. En analysant des milliers de clichés simulés, l’équipe a montré que ces blobs sont allongés, pas sphériques, et contiennent typiquement plusieurs nucléosomes entassés. Fait crucial, les contacts à l’intérieur de ces blobs correspondent étroitement aux blocs d’interaction de type domaine observés dans les données expérimentales, indiquant que les blobs ne sont pas des accidents aléatoires mais des unités 3D fondamentales de l’organisation de la chromatine. Les simulations prédisent même des frontières de domaine subtiles supplémentaires difficiles à détecter expérimentalement, suggérant que ce modèle physique peut révéler des caractéristiques fines cachées dans des données bruitées.
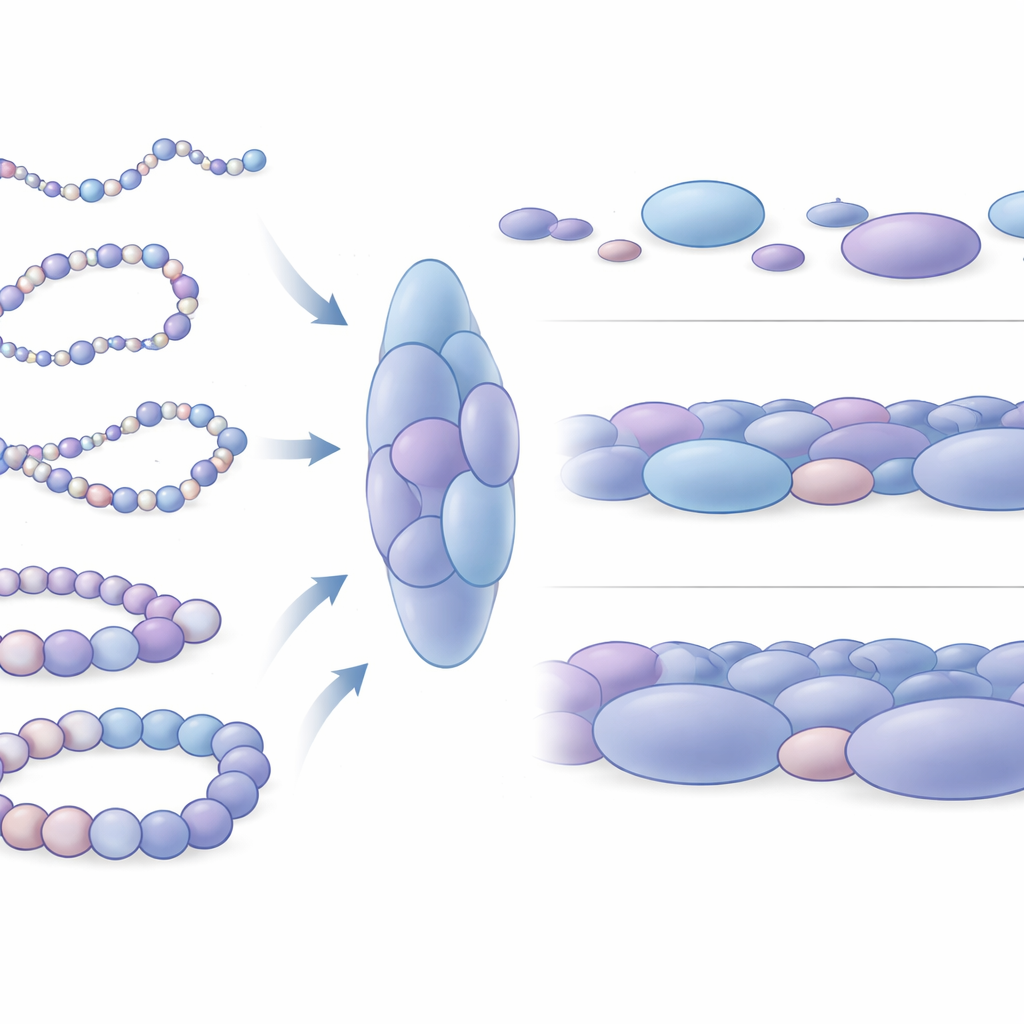
Comment les différences d’empilement reflètent l’activité génique
Les chercheurs ont ensuite demandé comment ces blobs diffèrent autour de gènes actifs par rapport à des gènes silencieux. Ils se sont concentrés sur quatre régions humaines bien étudiées, incluant deux gènes impliqués dans le maintien des cellules souches dans un état flexible et indifférencié (Nanog et Lin28A) et deux gènes de contrôle du développement (HoxB4 et HoxA13) qui sont éteints dans ces mêmes cellules. Autour des gènes inactifs, les blobs étaient en moyenne plus grands et plus densément empaquetés, les nucléosomes formant des arrangements locaux plus fermés. En revanche, les blobs proches des gènes actifs étaient plus petits, un peu plus lâches et plus variables. À une échelle plus large, l’ADN autour des gènes actifs explorait bien plus de formes distinctes et était mécaniquement plus flexible, tandis que les régions entourant les gènes silencieux se comportaient comme des segments de chromatine plus rigides. Cette différence mécanique affecte probablement la facilité avec laquelle des éléments régulateurs distants de l’ADN peuvent se rencontrer et coopérer avec les interrupteurs géniques.
Pourquoi cela compte pour comprendre le contrôle des gènes
Dans l’ensemble, les résultats dessinent un tableau où le génome est composé d’amas dynamiques de nucléosomes dont la taille, la forme et l’espacement contribuent à déterminer si les gènes voisins sont accessibles ou verrouillés. Le nouveau modèle relie les données expérimentales de contacts, les cartes de nucléosomes et des principes physiques dans un cadre unique qui explique comment les gènes de cellules souches peuvent rester flexibles et interactifs tandis que les gènes de développement restent enfermés dans des voisinages plus compacts et rigides. Pour les non-spécialistes, l’idée clé est que l’activité génique n’est pas régie par la seule séquence d’ADN ; elle dépend aussi de la manière dont cet ADN est replié en structures tridimensionnelles. En révélant les blobs de nucléosomes comme unités de base de ce repliement, ce travail offre une voie puissante pour relier l’architecture microscopique du génome à des processus à grande échelle comme le développement, l’identité cellulaire et la maladie.
Citation: Mittal, R., Heermann, D.W. & Bhattacherjee, A. An experimentally-informed polymer model reveals high resolution organization of genomic loci. Nat Commun 17, 2338 (2026). https://doi.org/10.1038/s41467-026-68928-w
Mots-clés: repliement de la chromatine, agrégats de nucléosomes, organisation 3D du génome, régulation des gènes, modélisation polymère