Clear Sky Science · fr
Un peptide cryptique neurotoxique issu de l’épissage cryptique de PKN1 dépendant de TDP-43
Menaces cachées à l’intérieur des cellules cérébrales
De nombreuses maladies cérébrales, dont la sclérose latérale amyotrophique (SLA) et la maladie d’Alzheimer, impliquent des agrégats d’une protéine appelée TDP-43. Les chercheurs savent que lorsque cette protéine cesse de fonctionner correctement, les neurones perdent des messages essentiels et finissent par mourir. Cette étude révèle une tournure plus surprenante : la défaillance de TDP-43 peut aussi amener les cellules cérébrales à fabriquer un mini‑protéine toxique jusque‑là inconnue, qui endommage à son tour les circuits de la mémoire. Comprendre ce protagoniste caché pourrait ouvrir de nouvelles voies pour le diagnostic et le traitement de démences dévastatrices.
Comment un relecteur cellulaire maintient l’ARN en ordre
À l’intérieur des neurones, TDP-43 joue le rôle d’un correcteur pour l’ARN, ces messages intermédiaires situés entre l’ADN et les protéines. Il se lie à de courtes séquences spécifiques et bloque l’insertion de « morceaux supplémentaires » errants dans ces messages. Lorsque TDP-43 est perdu ou mal localisé, comme dans la SLA et de nombreux cas d’Alzheimer, ces morceaux supplémentaires — appelés exons cryptiques — peuvent s’insérer dans l’ARN. Jusqu’à présent, la plupart des exons cryptiques connus provoquaient simplement une perte de la protéine normale en rendant le message instable et rapidement détruit. Il n’était pas clair que de tels événements puissent aussi générer de nouvelles protéines nocives.
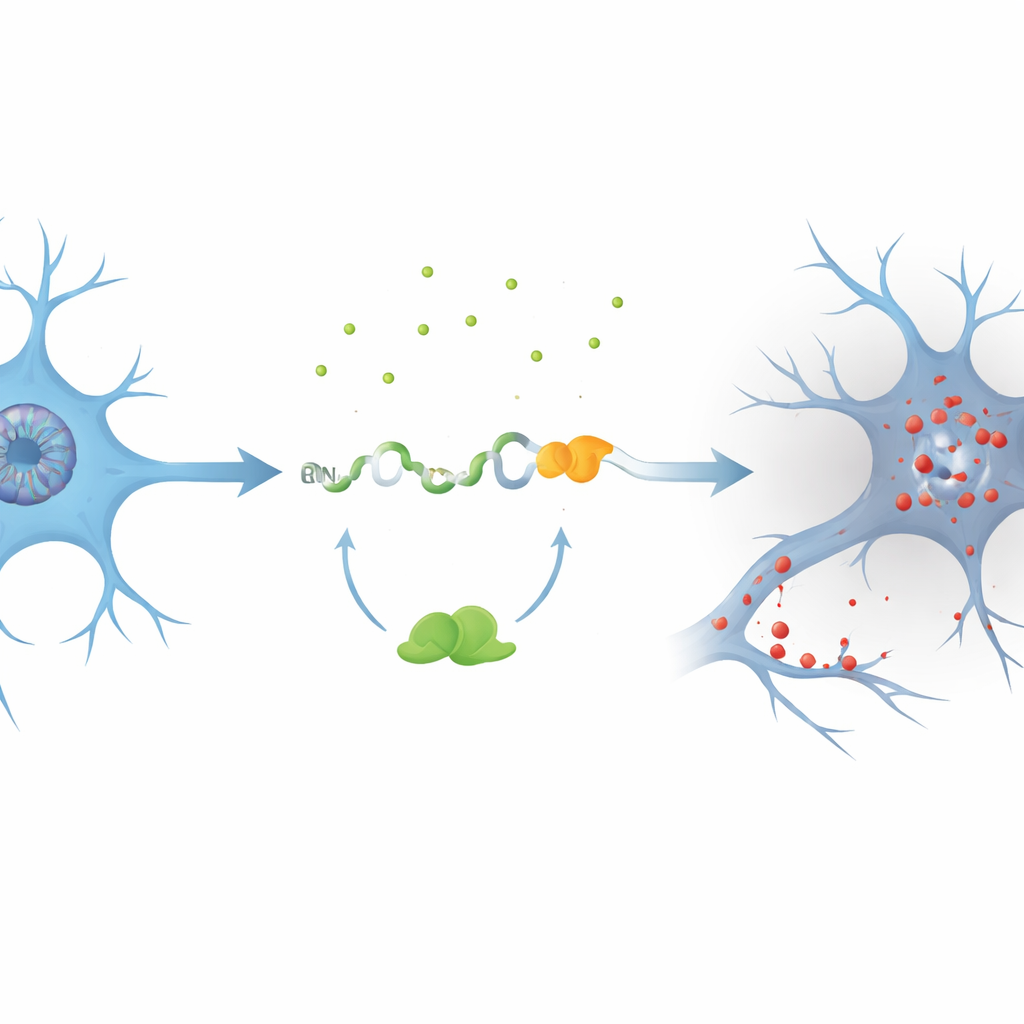
Un épissage cryptique crée un fragment toxique
Les auteurs se sont concentrés sur un gène nommé PKN1, qui contribue à maintenir l’ossature interne et la circulation des signaux des neurones. En utilisant des modèles cellulaires où TDP-43 était réduit, ils ont découvert un exon cryptique jusqu’alors non reconnu, baptisé PKN1‑5a1, inséré entre deux segments normaux de l’ARN de PKN1. Cette insertion introduit un signal d’arrêt précoce, produisant un ARN raccourci. Remarquablement, une partie de ce message défectueux échappe au système de contrôle qualité de la cellule et est traduite en un fragment stable de la protéine PKN1 contenant seulement ses 207 premiers acides aminés. L’équipe a nommé ce produit tronqué PKN207. Ils ont montré que TDP-43 empêche normalement cette erreur en se liant à plusieurs régions riches en UG flanquant l’exon cryptique ; quand cette liaison est perdue, l’exon est épissé et PKN207 est produit.
Preuves provenant de cerveaux de patients et de grandes bases de données
Pour savoir si cet événement se produit dans la maladie humaine, les chercheurs ont exploité des données de séquençage d’ARN provenant de centaines d’échantillons cérébraux et médullaires de patients atteints de SLA. Ils ont trouvé une activation répandue de l’exon cryptique PKN1‑5a1 dans des régions connues pour être touchées par la pathologie de TDP-43, comme le cortex moteur et la moelle épinière, mais pas dans le cervelet relativement épargné. Ils ont ensuite généré des anticorps hautement spécifiques qui reconnaissent uniquement la queue unique de PKN207, et non la protéine PKN1 pleine longueur. Dans des tissus hippocampiques de patients Alzheimer présentant également une TDP-43 anormale et phosphorylée, ces anticorps ont détecté une bande distincte correspondant à PKN207, tandis qu’une telle bande était absente dans les cerveaux témoins. Des ensembles de données Alzheimer supplémentaires ont confirmé que l’exon cryptique est activé dès les premiers stades de la maladie, suggérant que cette erreur moléculaire peut commencer bien avant l’apparition évidente des symptômes.
Une mini‑protéine avec un impact majeur sur la mémoire
La détection de PKN207 dans le cerveau malade humain posait la question clé : est‑il nocif ? Pour le tester, l’équipe a utilisé des virus pour forcer la production soit de PKN1 normal, soit de PKN207 spécifiquement dans l’hippocampe — une région cérébrale cruciale pour la mémoire — de souris jeunes. Des mois plus tard, les deux groupes de souris présentaient une altération de l’apprentissage dans le labyrinthe aquatique de Morris, nageant plus longtemps pour trouver une plateforme cachée. Leur liquide cérébro‑spinal contenait des niveaux plus élevés d’une protéine structurelle, la chaîne légère des neurofilaments, un marqueur de lésion axonale. En culture de neurones, l’augmentation de PKN207 a déclenché des lésions cellulaires, mesurées par la fuite d’une enzyme signalant une atteinte membranaire. Un profilage protéique détaillé de l’hippocampe a révélé des modifications étendues des voies liées à la force synaptique (potentialisation à long terme) et à des maladies neurodégénératives bien connues, avec une perturbation particulièrement marquée des molécules soutenant une signalisation efficace et un échafaudage d’axones sain.
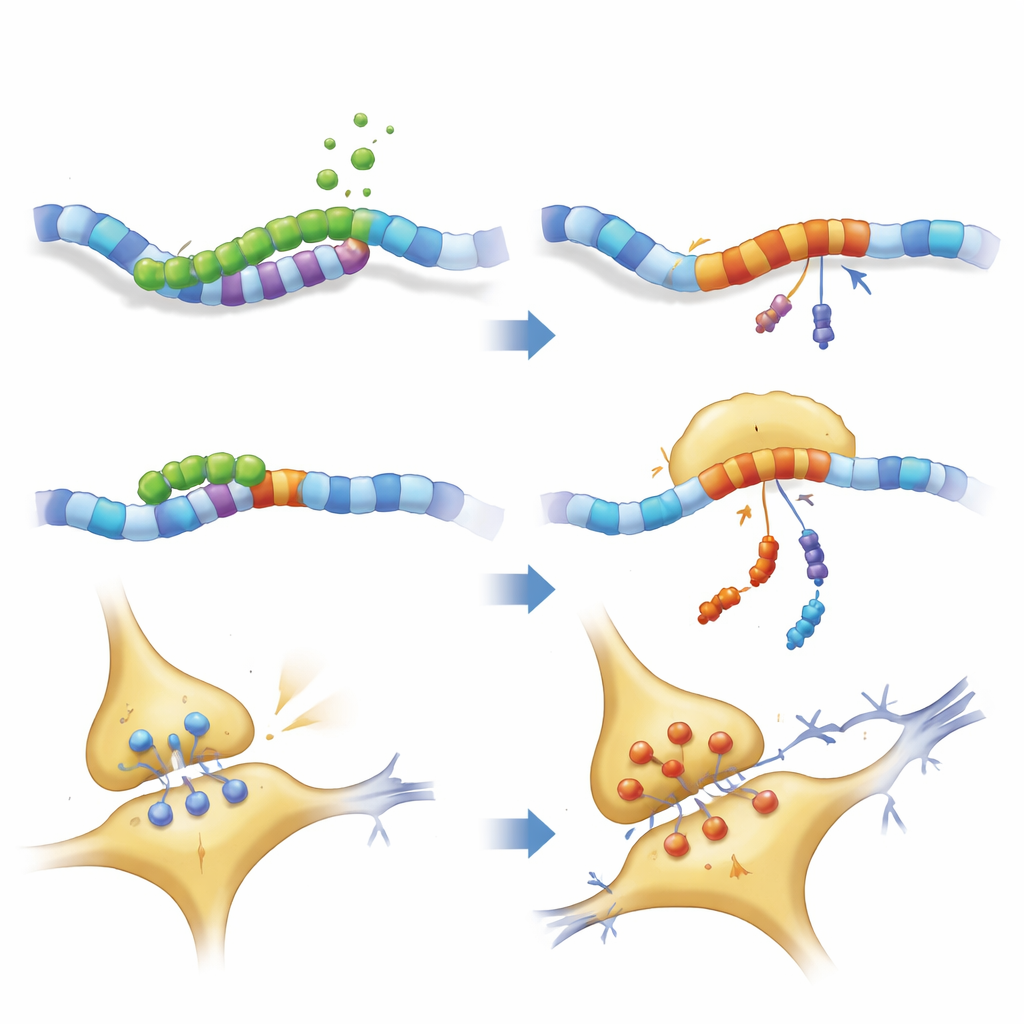
Comment le fragment perturbe le câblage cérébral
Un examen plus approfondi de la structure neuronale a montré que tant PKN1 pleine longueur que PKN207 perturbaient le réseau de neurofilaments qui donne aux axones leur forme et aide au transport des cargaisons. Les protéines motrices et d’échafaudage clés étaient réduites, tandis que certains composants des neurofilaments s’accumulaient, suggérant des embouteillages et un possible agrégat. Des enregistrements électriques de tranches d’hippocampe ont confirmé que les souris exprimant PKN207 présentaient une potentialisation à long terme affaiblie — le processus par lequel les synapses se renforcent après une activité répétée et qui constitue une base cellulaire largement acceptée de l’apprentissage et de la mémoire. Même si PKN207 ne possède pas le domaine enzymatique de PKN1, sa présence suffisait à imiter et parfois dépasser les effets perturbateurs de la protéine complète, ce qui implique que la région N‑terminale partagée peut, à elle seule, interférer avec l’homéostasie neuronale.
Pourquoi cette découverte est importante pour les maladies cérébrales
Ce travail ajoute une nouvelle couche à notre compréhension des troubles liés à TDP-43. Plutôt que de simplement provoquer la perte d’ARN essentiels, la défaillance de TDP-43 peut aussi engendrer un micro‑protéine stable et toxique qui sape les synapses et la cognition. L’exon cryptique PKN1‑5a1 et son produit peptidique PKN207 se distinguent désormais comme des biomarqueurs potentiels d’une dysfonction précoce de TDP-43 et comme cibles candidates pour des thérapies visant à corriger l’épissage ou à bloquer le fragment nocif. Plus largement, l’étude suggère que d’autres exons cachés pourraient également donner naissance à des peptides responsables de la maladie, orientant les chercheurs vers un paysage riche — et jusqu’ici négligé — de coupables moléculaires dans la neurodégénérescence.
Citation: Yang, M., Wang, Q., Yan, R. et al. A neurotoxic cryptic peptide arising from TDP-43-dependent cryptic splicing of PKN1. Nat Commun 17, 2963 (2026). https://doi.org/10.1038/s41467-026-68916-0
Mots-clés: TDP-43, épissage cryptique, PKN1, neurodégénérescence, Alzheimer et SLA