Clear Sky Science · fr
Modélisation de la fibrose pulmonaire à partir de cellules iPS humaines révèle que l'inhibition de p300/CBP supprime l'état cellulaire alvéolaire transitionnel
Pourquoi les poumons cicatrisés nous concernent tous
La fibrose pulmonaire idiopathique (FPI) est une maladie implacable dans laquelle les poumons se transforment progressivement en tissu cicatriciel, rendant chaque respiration plus difficile. Les médicaments actuels ne font que ralentir ce processus et provoquent souvent des effets secondaires gênants. Cette étude utilise des outils de pointe en biologie des cellules souches et en génomique pour recréer des poumons cicatrisés en laboratoire, posant une question simple mais essentielle : peut‑on identifier un interrupteur qui éloigne les cellules pulmonaires endommagées d’un état néfaste et les ramène vers la réparation ?
Une fenêtre cultivée en laboratoire sur un poumon cicatrisé
Pour étudier la FPI, les chercheurs ont construit des mini‑poumons à partir de cellules souches pluripotentes induites humaines (iPSC). Ces iPSC ont été guidées pour devenir des cellules alvéolaires — les cellules qui tapissent les minuscules sacs d’air où l’oxygène passe dans le sang — et ont été cultivées en présence de fibroblastes pulmonaires, les cellules du tissu conjonctif qui forment la cicatrice. Enrobés dans un gel mou, ces « organoïdes alvéolaires » se comportaient comme du tissu pulmonaire réel. Lorsqu’ils ont été exposés au médicament de chimiothérapie bléomycine, un déclencheur connu de lésion pulmonaire, les gels se sont contractés sous l’effet de tractions exercées par les fibroblastes, reproduisant la contraction tissulaire observée dans la fibrose.
Avec ce système, l’équipe a criblé une bibliothèque de 264 petites molécules et a mesuré automatiquement dans quelle mesure chaque composé empêchait la contraction du gel, s’appuyant sur un outil d’analyse d’images par apprentissage profond pour obtenir des mesures objectives. Beaucoup de composés n’avaient aucun effet, mais une famille s’est nettement distinguée : les inhibiteurs des protéines p300 et CBP, qui participent au contrôle de la façon dont l’ADN est emballé et des gènes qui sont activés. Les huit composés ciblant p300/CBP dans la bibliothèque ont réduit la contraction à faibles doses, mettant en évidence cette voie comme une cible prometteuse contre la fibrose. 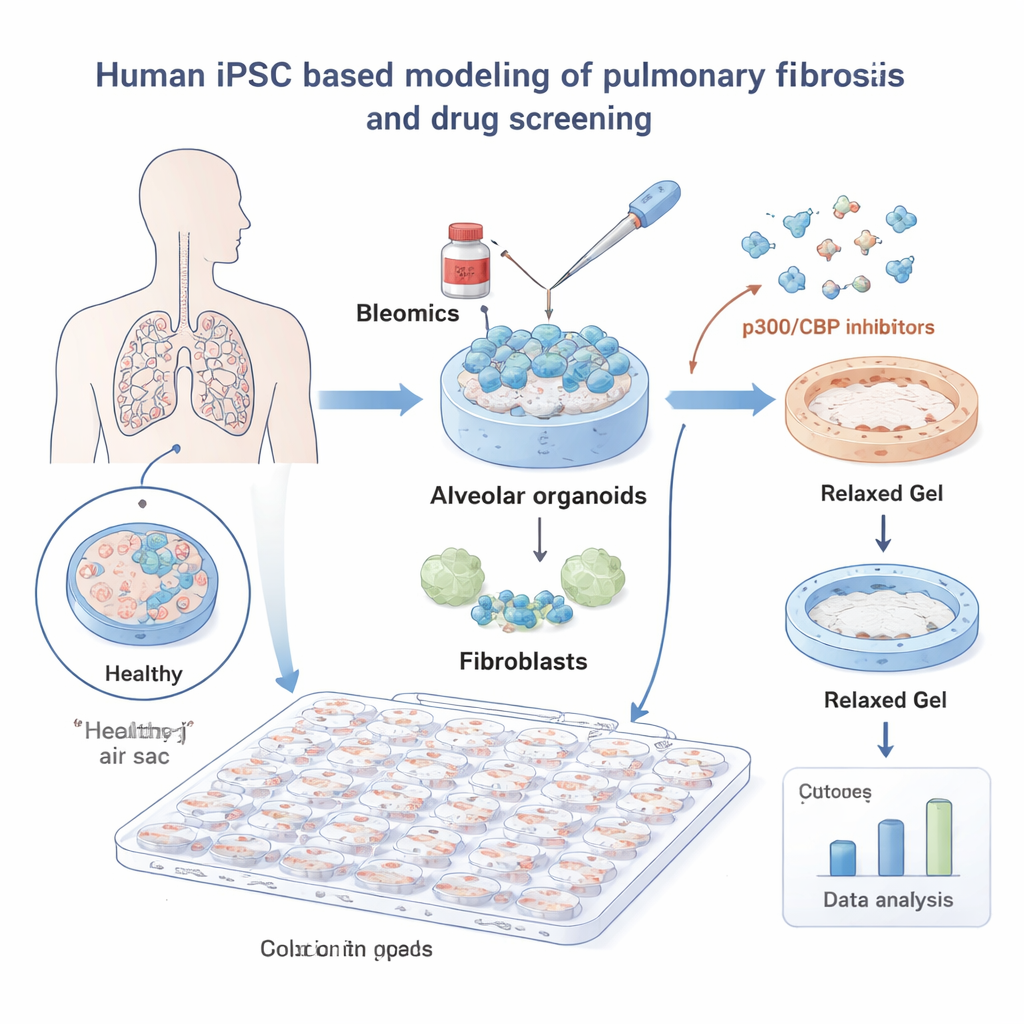
Les fauteurs de trouble : les cellules pulmonaires transitionnelles
Des travaux récents ont mis au jour un type cellulaire « intermédiaire » problématique dans les poumons malades, appelé état cellulaire alvéolaire transitionnel. Normalement, des cellules de soutien appelées cellules AT2 maturent en cellules AT1 ultras‑minces qui recouvrent les alvéoles et permettent les échanges gazeux. Dans la FPI, cependant, les cellules AT2 restent souvent bloquées dans cet état transitionnel, exprimant des gènes de stress et de réparation tout en n’achevant pas leur transformation en cellules AT1 pleinement fonctionnelles. Ces cellules transitionnelles s’accumulent dans les régions fibreuses et communiquent fortement avec les fibroblastes, mais il n’était pas clair si elles étaient simplement un sous‑produit des lésions ou des acteurs actifs de la cicatrisation.
En séquençant l’ARN et en profileant la chromatine ouverte dans leurs organoïdes, les auteurs ont montré que les cellules transitionnelles induites dans leur modèle correspondaient étroitement à celles trouvées dans les poumons de patients atteints de FPI. Ces cellules transitionnelles présentaient des signatures géniques de stress, d’inflammation et de remodelage de la matrice, et elles activaient fortement les fibroblastes pulmonaires adultes mis en coculture. De façon cruciale, lorsque p300/CBP était bloqué, les marqueurs de l’état transitionnel diminuaient, l’identité AT2 était mieux préservée et l’activation des fibroblastes faiblissait. Autrement dit, les médicaments ne tuaient pas les cellules de façon générale ; ils empêchaient sélectivement les cellules AT2 d’être piégées dans ce limbe néfaste.
Démêler les commutateurs moléculaires
Pour comprendre comment p300/CBP oriente cette décision de destin, l’équipe a examiné des marques chimiques sur les histones — les protéines qui aident à empaqueter l’ADN. Une marque particulière, l’acétylation de H3K27, est couramment déposée par p300/CBP aux enhancers et promoteurs actifs. Dans les cellules transitionnelles, des régions proches de gènes de réponse au stress et pro‑fibrotiques portaient une forte acétylation de H3K27 et étaient enrichies en sites de liaison pour des facteurs de transcription tels qu’AP‑1 et HNF1B. Lorsque les cellules étaient traitées par des inhibiteurs de p300/CBP, ces marques d’acétylation diminuaient à ces loci, et l’expression de nombreux gènes pro‑fibrotiques chutait. Bloquer AP‑1 directement, ou réduire AP‑1 et HNF1B avec de petits ARN interférents, limitait de même le programme transitionnel et la contraction des organoïdes, reliant ainsi ce trio — p300/CBP, AP‑1 et HNF1B — au moteur qui alimente le remodelage fibrotique.
Au‑delà de la boîte de culture, l’étude a testé un des inhibiteurs, CBP30, chez des souris avec une lésion pulmonaire induite par bléomycine. Les animaux traités par CBP30 présentaient moins de cellules épithéliales transitionnelles, une moindre activation des myofibroblastes formateurs de cicatrice et une expression réduite des marqueurs de fibrose. Cette validation croisée entre modèles humains de cellules souches et un modèle animal renforce l’idée que p300/CBP n’est pas qu’un artefact de laboratoire mais bien un régulateur réel de la cicatrisation pulmonaire. 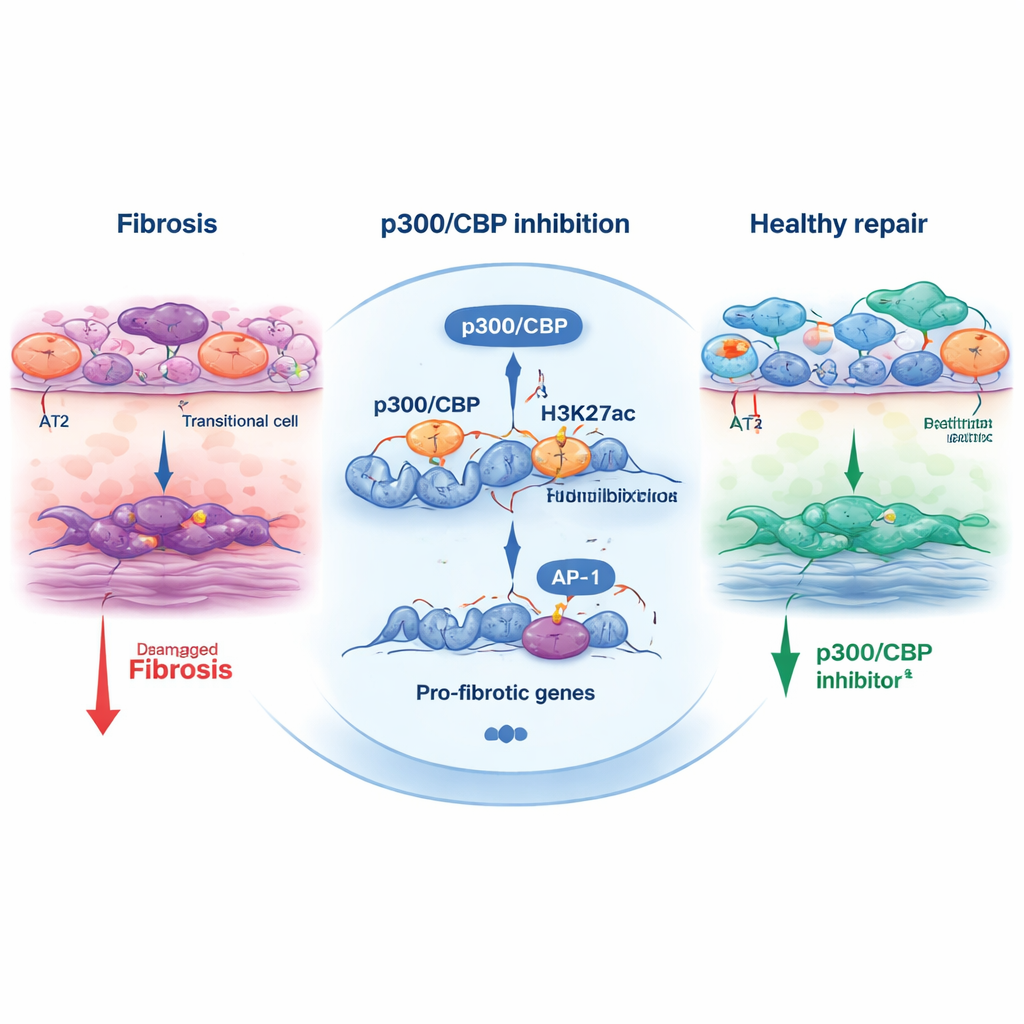
Ce que cela signifie pour les traitements futurs
Pour les non‑spécialistes, la conclusion essentielle est que les auteurs ont construit un modèle humain réaliste de poumons fibreux et l’ont utilisé pour mettre en lumière une nouvelle cible médicamenteuse. Leur travail suggère que la cicatrisation pulmonaire est en partie conduite par un état cellulaire transitionnel réversible et induit par le stress qui désoriente le tissu environnant. En réduisant l’activité de p300/CBP, il pourrait être possible d’apaiser cet état, de maintenir les cellules alvéolaires sur une trajectoire de développement saine et de diminuer les signaux qui poussent les fibroblastes à s’emballer. Si les inhibiteurs de p300/CBP doivent encore être optimisés pour la sécurité et testés en clinique, cette étude oriente vers des thérapies qui s’attaquent à la mauvaise communication cellulaire à la racine de la FPI plutôt que de se contenter d’en ralentir les conséquences.
Citation: Tsutsui, Y., Masui, A., Konishi, S. et al. Human iPSC-based Modeling of Pulmonary Fibrosis Reveals p300/CBP Inhibition Suppresses Alveolar Transitional Cell State. Nat Commun 17, 1214 (2026). https://doi.org/10.1038/s41467-026-68909-z
Mots-clés: fibrose pulmonaire idiopathique, organoïdes alvéolaires, inhibiteurs p300/CBP, cellules épithéliales transitionnelles, cellules souches pulmonaires