Clear Sky Science · fr
Immunothérapie anti-TLR2 module la propagation neuronale vers oligodendrocyte de l’α-synucléine dans des modèles murins et humains
Pourquoi cette recherche est importante
L’atrophie multisystématisée (AMS) est une maladie cérébrale rare mais rapidement évolutive qui combine des troubles du mouvement de type parkinsonien avec des déficits de l’équilibre et du système autonome, comme des chutes de tension. Les médecins peuvent soulager les symptômes, mais ils ne peuvent actuellement pas ralentir la progression de la maladie. Cette étude révèle comment une protéine mal repliée se transmet entre cellules cérébrales pour endommager « l’isolation » des circuits nerveux, et montre qu’un traitement par anticorps ciblé peut interrompre ce processus dans des modèles cellulaires et animaux. Ce travail indique une stratégie pharmaceutique concrète qui pourrait un jour modifier la trajectoire de l’AMS plutôt que de se limiter à traiter ses symptômes.
Comment le câblage cérébral se dérègle
Dans de nombreux troubles du mouvement, y compris la maladie de Parkinson, des amas d’une protéine appelée alpha-synucléine s’accumulent à l’intérieur des neurones. Dans l’AMS, cependant, les amas les plus marquants se forment à l’intérieur de cellules de soutien appelées oligodendrocytes, qui en temps normal enveloppent les fibres nerveuses d’une gaine grasse de myéline accélérant la conduction électrique. Étonnamment, les oligodendrocytes produisent très peu d’alpha-synucléine eux-mêmes, laissant une énigme ancienne : d’où proviennent les dépôts massifs de protéine dans ces cellules ? Les auteurs ont d’abord confirmé, à l’aide d’échantillons de cerveaux humains et d’analyses d’ARN à grande échelle, que les oligodendrocytes produisent effectivement beaucoup moins d’alpha-synucléine que les neurones, renforçant l’hypothèse que la protéine nocive doit provenir de l’extérieur.
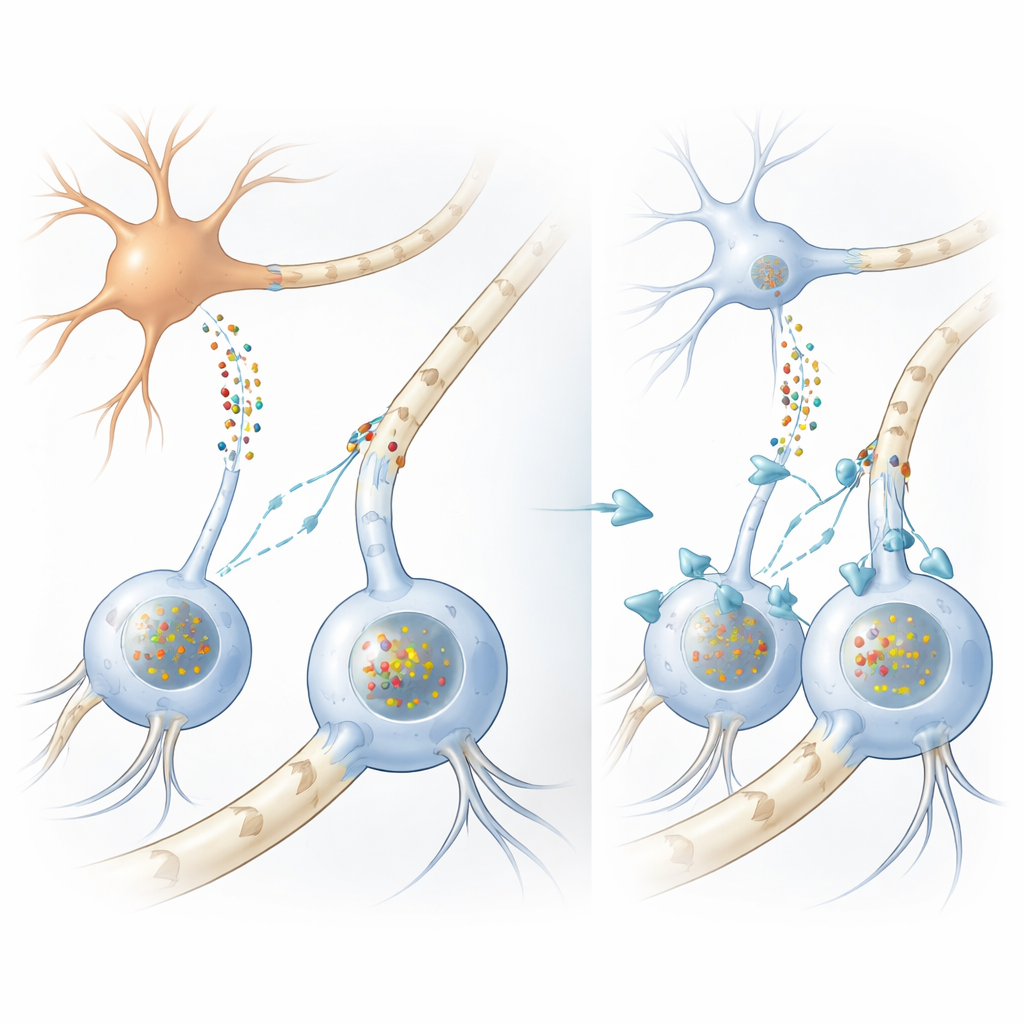
La protéine transférée du neurone à la cellule de soutien
Pour tester cela, l’équipe a mis au point plusieurs modèles complémentaires. En culture, ils ont fait croître des cellules humaines ressemblant à des oligodendrocytes à partir de cellules souches neurales et les ont exposées au milieu prélevé sur des cellules de type neuronal génétiquement modifiées pour libérer de grandes quantités d’alpha-synucléine. Les cellules de soutien ont internalisé cette protéine et développé des amas très semblables aux inclusions gliales observées dans les cerveaux d’AMS, avec les mêmes marques chimiques et protéines auxiliaires. Lorsque les chercheurs ont examiné une lignée de souris produisant de l’alpha-synucléine humaine mutante uniquement dans les neurones, ils ont de nouveau trouvé des amas de protéine humaine à l’intérieur des oligodendrocytes dans la matière blanche, bien que ces cellules n’exprimaient pas le gène humain. Ensemble, ces expériences montrent que l’alpha-synucléine peut passer des neurones aux oligodendrocytes et y former des inclusions semblables à celles observées en maladie.
La porte située à la surface cellulaire
Ensuite, les scientifiques se sont demandé comment la protéine entre dans les oligodendrocytes. Des travaux antérieurs avaient identifié le senseur immunitaire Toll-like receptor 2 (TLR2) à la surface des cellules comme site d’accrochage de l’alpha-synucléine dans les neurones et les microglies. En exploitant des jeux de données d’expression génique issus de cerveaux d’AMS, l’équipe a constaté que les oligodendrocytes des patients présentaient des niveaux de TLR2 anormalement élevés par rapport aux témoins, et qu’un TLR2 plus élevé était associé à des niveaux plus faibles de gènes liés à la myéline, tels que la protéine basique de la myéline. Cette relation n’apparaissait pas dans plusieurs jeux de données indépendants sur la maladie de Parkinson, laissant penser que la sensibilité des oligodendrocytes à l’alpha-synucléine via TLR2 est une caractéristique distinctive de l’AMS plutôt qu’un trait général de tous les troubles à synucléine.
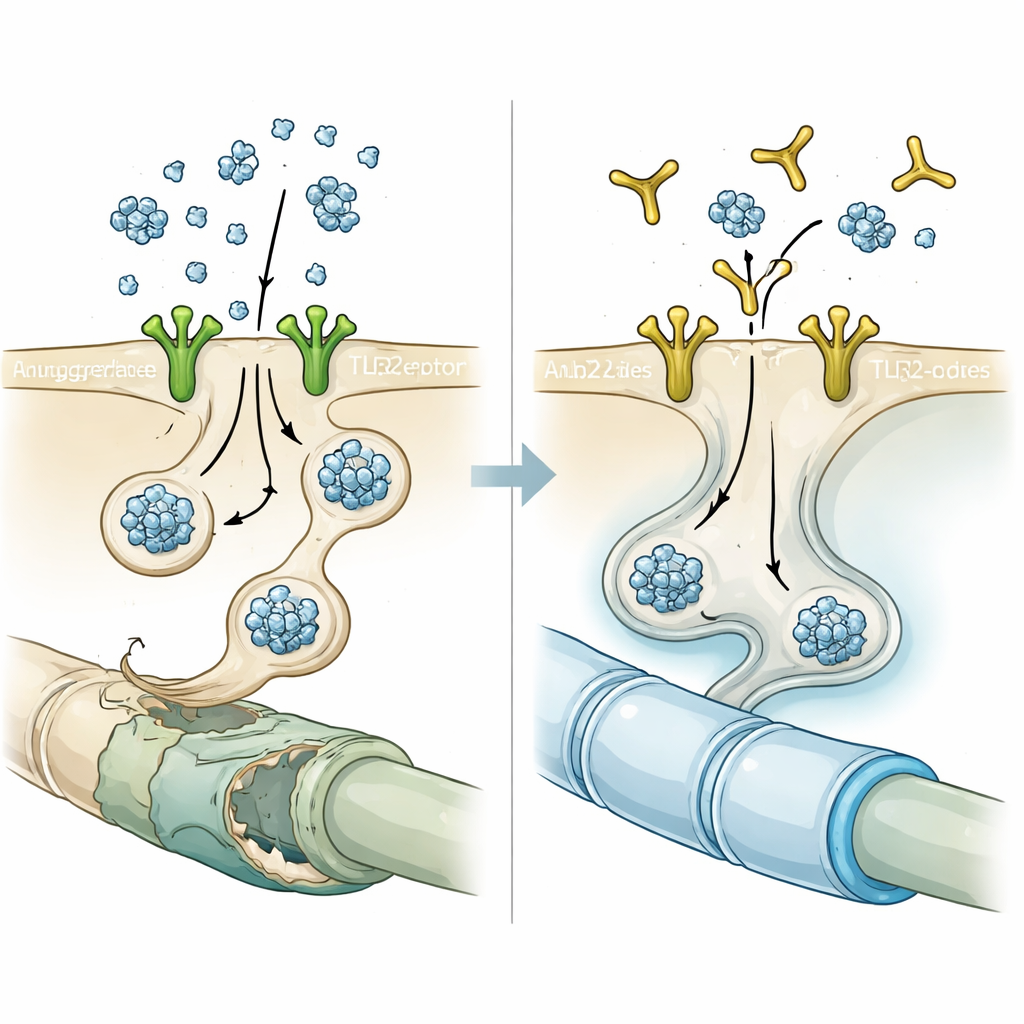
Bloquer la porte avec un anticorps
Fort de cet indice, les chercheurs ont testé NM-101, un anticorps conçu pour se fixer à TLR2 et empêcher son activation. En cultures cellulaires, un prétraitement bref des oligodendrocytes par NM-101 avant l’ajout d’alpha-synucléine d’origine neuronale a fortement réduit le nombre et l’intensité des amas de type inclusion. Chez des souris soit surexprimant l’alpha-synucléine neuronale soit ayant reçu des injections de fibrilles d’alpha-synucléine préformées, des perfusions hebdomadaires de NM-101 ont diminué les protéines agrégées dans la matière blanche, atténué les réponses inflammatoires des microglies et des astrocytes, et réduit l’activation d’une enzyme inflammatoire appelée caspase-1 à l’intérieur des oligodendrocytes. Les animaux traités ont vécu plus longtemps et obtenu de meilleurs résultats aux tests moteurs, suggérant que les effets protecteurs de l’anticorps étaient sur le plan fonctionnel significatifs et non de simples curiosités microscopiques.
Réparer l’isolation endommagée
Parce que les oligodendrocytes fabriquent la myéline du cerveau, l’équipe a examiné si le transfert d’alpha-synucléine endommageait la myéline et si le blocage de TLR2 pouvait aider. Le séquençage ARN monocellulaire d’oligodendrocytes d’origine humaine exposés à de l’alpha-synucléine conditionnée par des neurones a révélé un large basculement loin d’un état mature producteur de myéline vers un profil plus immature, de type progéniteur, avec de nombreux gènes clés de la myéline réprimés. Des études d’expression génique parallèles sur des oligodendrocytes isolés au laser provenant de patients AMS et du modèle murin ont montré une signature partagée : une expression réduite des gènes impliqués dans la formation et le maintien de la myéline. Au microscope électronique, la matière blanche chez les souris exprimant l’alpha-synucléine présentait des gaines de myéline plus minces et désorganisées. Le traitement par NM-101 a inversé beaucoup de ces changements, épaississant la myéline, restaurant les niveaux de protéines de la myéline et normalisant l’expression des gènes nécessaires à la maturation des oligodendrocytes.
Ce que cela signifie pour les traitements futurs
L’étude soutient un scénario clair : dans l’AMS, l’alpha-synucléine produite par les neurones peut se répandre dans les oligodendrocytes via TLR2 à leur surface, s’y accumuler, déclencher l’inflammation, perturber le programme de développement des cellules et éroder la gaine de myéline des connexions cérébrales. En bloquant TLR2 avec un anticorps ciblé, les chercheurs ont pu interrompre cette chaîne d’événements dans des modèles murins et cellulaires humains, réduisant les inclusions toxiques, apaisant l’inflammation, réparant la myéline et améliorant la survie et la motricité. Bien que NM-101 doive encore être rigoureusement testé chez l’humain, ce travail établit le transfert de protéine dépendant de TLR2 comme un moteur central de la pathologie de type AMS et met en avant l’immunothérapie anti-TLR2 comme une stratégie prometteuse pour ralentir ou prévenir cette maladie dévastatrice.
Citation: Bae, EJ., Ham, S., Jeong, Y.W. et al. Anti-TLR2 immunotherapy modulates neuron-to-oligodendrocyte propagation of α-synuclein in mouse and human models. Nat Commun 17, 2175 (2026). https://doi.org/10.1038/s41467-026-68870-x
Mots-clés: atrophie multisystématisée, alpha-synucléine, oligodendrocytes, dégâts de la myéline, immunothérapie