Clear Sky Science · fr
Un atlas de quantification absolue des petits ARN non codants à travers divers tissus de mammifères et lignées cellulaires
Pourquoi les très petits ARN comptent
À l’intérieur de chaque cellule, des flottes de minuscules molécules d’ARN contribuent à décider quels gènes sont activés ou réprimés. Ces petits ARN non codants agissent comme des variateurs pour nos programmes génétiques, façonnant le développement, le fonctionnement des organes et les maladies. Pourtant, malgré des technologies de séquençage puissantes, les scientifiques ont eu du mal à mesurer précisément combien de ces molécules sont présentes dans différents types cellulaires et tissus. Cette étude présente une méthode plus exacte pour les compter et construit un atlas détaillé qui montre leur abondance réelle à travers de nombreux tissus de mammifères et lignées cellulaires courantes en laboratoire.
Une méthode plus claire pour compter les petits ARN
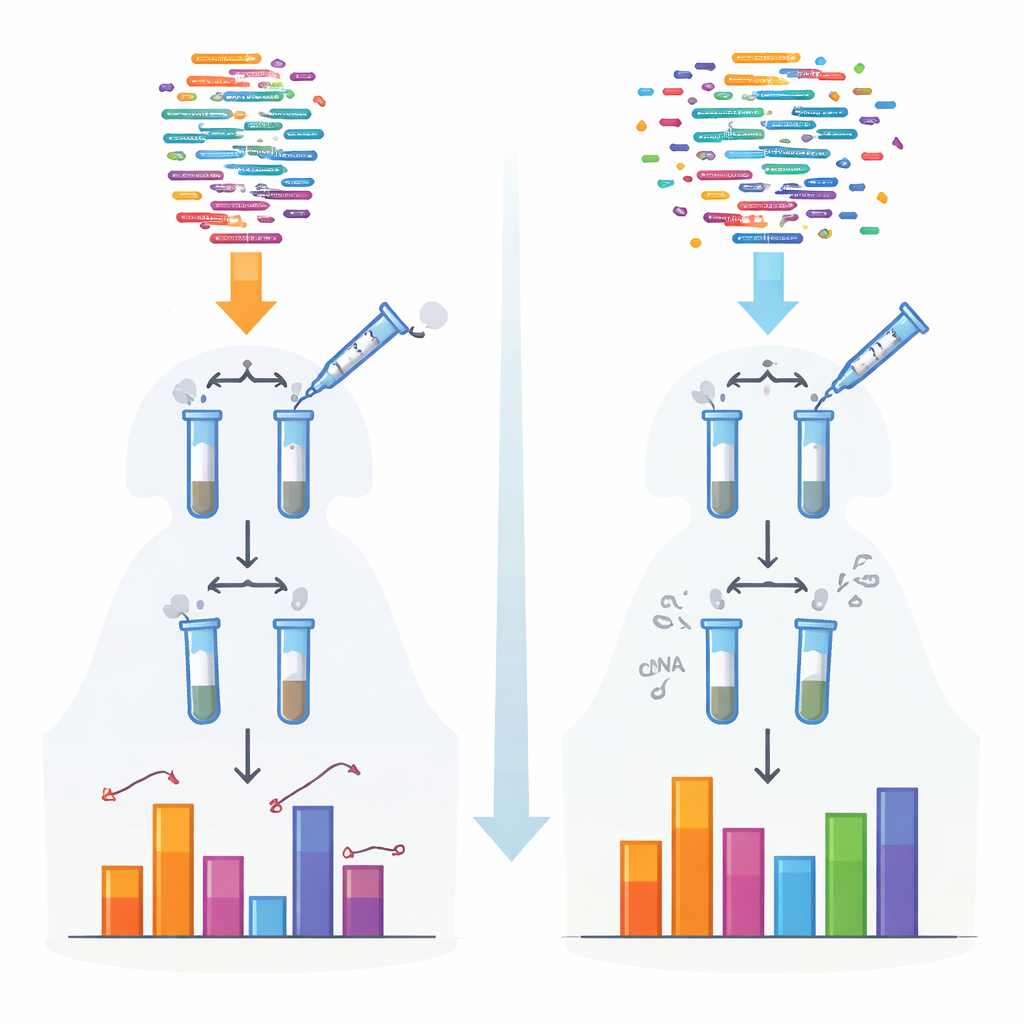
Les méthodes traditionnelles de séquençage des petits ARN reposent sur des enzymes qui fixent des adaptateurs avant la lecture des molécules. Ces enzymes privilégient certaines formes et terminaisons chimiques, de sorte que certains ARN sont capturés efficacement tandis que d’autres sont manqués ou sous‑estimés. Ce biais est particulièrement marqué pour des classes spécifiques comme les piARN et les petits ARN végétaux, qui portent des coiffes chimiques protectrices à leurs extrémités. Les auteurs ont créé un nouveau protocole, nommé 4NBoost, qui re‑conçoit les adaptateurs et les conditions de réaction pour lisser ces préférences et ajoute des codes-barres moléculaires intégrés permettant de distinguer les molécules réelles des copies générées lors de l’amplification.
Transformer un protocole en outil de mesure
Pour convertir 4NBoost d’un signal relatif en un véritable outil de mesure, l’équipe a ajouté des ARN synthétiques « spike‑in » conçus avec soin à des concentrations connues couvrant une large plage. En comparant combien de fois chaque spike‑in était lu par le séquenceur avec la quantité initialement ajoutée, ils ont établi des courbes standard qui convertissent les nombres de lectures en nombres absolus de molécules. Des tests avec différents mélanges de spike‑in et des ARN témoins ajoutés ont montré que 4NBoost pouvait suivre l’abondance avec précision sur plusieurs ordres de grandeur, y compris pour des ARN portant des modifications chimiques problématiques. Même en partant d’aussi peu qu’un nanogramme d’ARN total, la méthode capturait fidèlement le paysage des petits ARN.
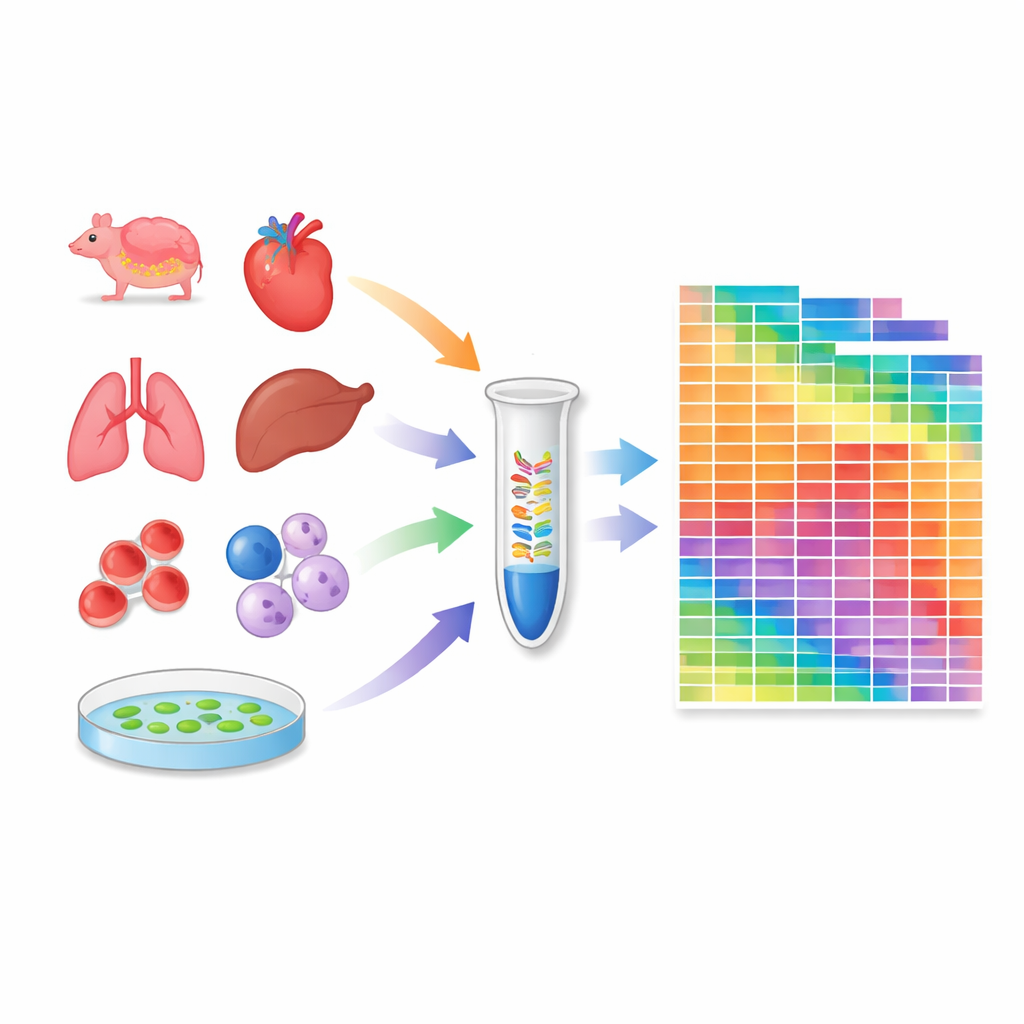
Construire un atlas à travers tissus et lignées cellulaires
Equipés de ce protocole calibré, les chercheurs ont profilé 259 échantillons : 20 tissus de souris, 18 de macaques crabiers, 24 lignées cellulaires humaines et murines largement utilisées, et plusieurs tissus de la plante modèle Arabidopsis. Pour chaque échantillon, ils ont estimé le nombre absolu de molécules pour des milliers de microARN et de piARN. Cela a révélé combien d’espèces de microARN différentes sont présentes dans chaque contexte et comment leurs quantités totales varient entre tissus et espèces. Certaines lignées cellulaires et organes possèdent des répertoires de microARN particulièrement riches, tandis que d’autres, comme les cellules sanguines, sont dominés par quelques espèces très abondantes. L’atlas a également mis en évidence des différences substantielles entre tissus de souris et de singe, soulignant que la régulation par petits ARN peut être spécifique à l’espèce.
Corriger les anciennes données et réviser des hypothèses courantes
Lorsque le nouvel atlas a été confronté aux bases de données de petits ARN populaires construites avec des méthodes conventionnelles, des discordances marquées sont apparues. Plusieurs familles importantes de microARN — telles que miR‑19 et miR‑29 — se sont révélées bien plus abondantes qu’on ne le pensait auparavant, tandis que d’autres — comme les familles très étudiées let‑7 et miR‑10 — étaient souvent surestimées. L’étude a aussi réexaminé laquelle des « branches » de chaque précurseur en épingle est réellement utilisée dans les cellules, découvrant des cas où les annotations actuelles indiquent la mauvaise chaîne principale. Pour sauver la richesse des jeux de données biaisés existants, les auteurs ont entraîné un modèle d’apprentissage automatique qui apprend comment les mesures conventionnelles dévient de 4NBoost puis les corrige mathématiquement pour mieux refléter les abondances réelles.
Une ressource publique pour explorer les petits ARN
Toutes les mesures 4NBoost et le modèle de correction sont mis librement à disposition via une plateforme en ligne appelée SmRNAQuant. Les chercheurs peuvent parcourir ou télécharger les niveaux absolus de petits ARN pour des tissus, des lignées cellulaires ou des microARN précis, et peuvent téléverser leurs propres données préparées avec un kit courant pour obtenir des valeurs corrigées des biais. Pour les non‑spécialistes, le message essentiel est que le comptage importe : de très petites différences dans le nombre de copies d’un petit ARN peuvent faire la différence entre une régulation génique active et l’absence d’effet. En fournissant des nombres plus fiables et un moyen de corriger les données anciennes, ce travail pose une base quantitative plus solide pour comprendre comment les petits ARN façonnent la biologie normale et la maladie.
Citation: Xiao, W., Zheng, Y., Zhang, H. et al. An absolute quantification atlas of small non-coding RNAs across diverse mammalian tissues and cell lines. Nat Commun 17, 2314 (2026). https://doi.org/10.1038/s41467-026-68812-7
Mots-clés: quantification des microARN, petits ARN non codants, biais du séquençage de l'ARN, atlas d'expression tissulaire, correction par apprentissage automatique