Clear Sky Science · fr
Évaluation des technologies d’atlasage single‑cell ATAC‑seq à l’aide de modèles séquence‑vers‑fonction
Lire le manuel d’instructions de la cellule
Toutes les cellules de votre corps lisent le même ADN, pourtant les cellules cérébrales, musculaires et immunitaires se comportent très différemment. Cet article traite d’un casse‑tête central derrière cette diversité : comment de courtes séquences d’ADN appelées enhancers fonctionnent comme des interrupteurs pour activer ou réprimer des gènes dans des types cellulaires spécifiques. Les auteurs montrent que de nouvelles technologies de laboratoire moins chères peuvent générer les jeux de données massifs nécessaires pour entraîner des modèles d’apprentissage profond modernes qui lisent les séquences d’ADN et prédisent quels enhancers sont actifs dans quelles cellules, nous rapprochant d’un véritable décodage de la « grammaire » régulatrice du génome.
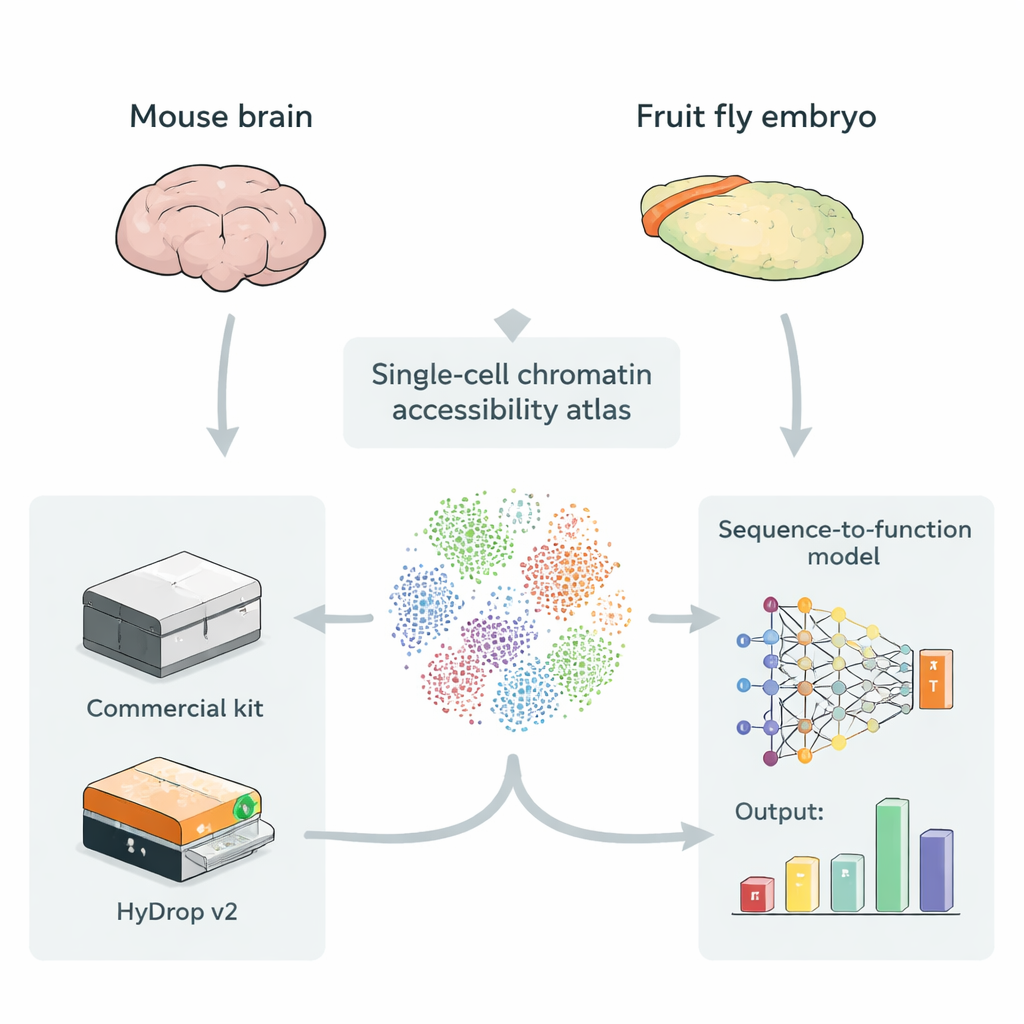
Cartographier l’ADN accessible dans des cellules individuelles
Les enhancers se situent généralement dans des régions d’ADN plus ouvertes et accessibles, ce qui facilite la liaison des protéines régulatrices. Une technique appelée single‑cell ATAC‑seq mesure quelles parties du génome sont ouvertes dans des milliers à des centaines de milliers de cellules individuelles simultanément, créant un « atlas » de l’ADN accessible à travers de nombreux types cellulaires. Ces atlas sont un carburant idéal pour des modèles d’apprentissage profond qui prennent la séquence d’ADN brute en entrée et apprennent à prédire à quel point chaque petite région fonctionne comme un enhancer dans chaque type cellulaire. Jusqu’à présent, cependant, la plupart de ces atlas reposaient sur des instruments commerciaux coûteux, soulevant la question de savoir si des méthodes open‑source à faible coût peuvent fournir des données d’entraînement de valeur équivalente pour ces modèles.
Une alternative open‑source aux plateformes commerciales
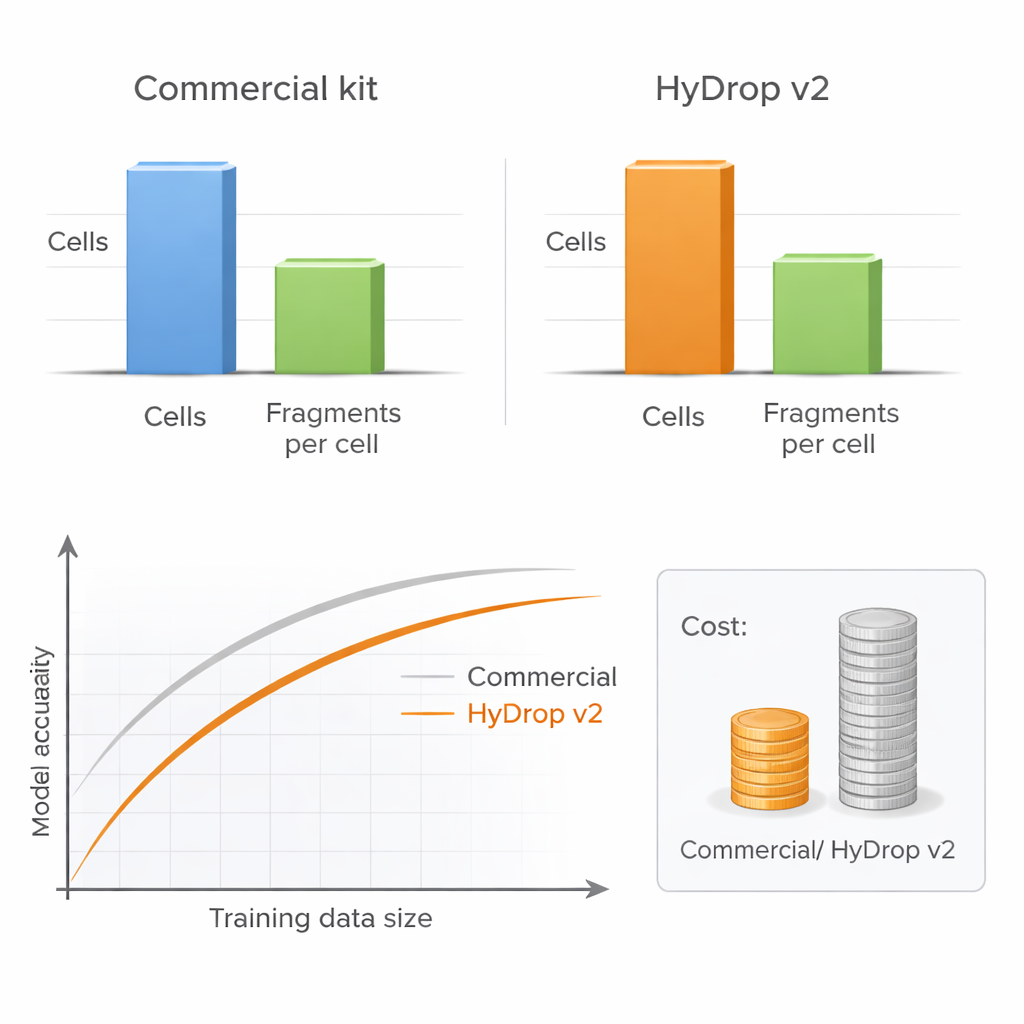
Les auteurs présentent HyDrop v2, une méthode améliorée basée sur des gouttelettes pour le single‑cell ATAC‑seq qui utilise des billes hydrogel personnalisées pour barcoder les cellules individuelles. Ils comparent HyDrop v2 à un kit commercial largement utilisé en construisant de grands atlas à partir de deux systèmes très différents : le cortex moteur de souris adulte et des embryons de drosophile en stade tardif. HyDrop v2 produit une qualité de données comparable — récupérant les mêmes types cellulaires majeurs et des ensembles de régions d’ADN accessibles très similaires — tout en coûtant environ quatorze fois moins par échantillon de cerveau de souris. Fait important, les données issues d’expériences HyDrop v2 s’intègrent harmonieusement avec les données commerciales, ce qui permet aux chercheurs de combiner les plateformes lors de la constitution de très grands atlas.
Entraîner des modèles d’apprentissage profond pour lire la logique des enhancers
Pour tester si des données moins coûteuses suffisent pour des modélisations avancées, l’équipe entraîne des modèles séquence‑vers‑fonction d’apprentissage profond sur des atlas issus soit de données commerciales soit de HyDrop v2. Ces modèles apprennent directement à partir de la séquence d’ADN à prédire l’accessibilité de chaque région dans chaque type cellulaire, et peuvent mettre en évidence de courts motifs de séquence qui correspondent probablement à des sites de liaison pour des protéines régulatrices spécifiques. Dans le cortex de souris, les modèles entraînés sur les données HyDrop v2 égalent les modèles basés sur des données commerciales en précision globale et en capacité à retrouver des « interrupteurs » d’enhancer connus qui avaient été validés in vivo. Chez l’embryon de drosophile, les deux plateformes permettent des modèles capables d’analyser des régions de 2 000 paires de bases et d’identifier les segments centraux d’environ 500 paires de bases qui pilotent réellement l’activité enhancer spécifique aux tissus, comme des régions contrôlant l’expression génique des neuroblastes ou des muscles.
Plus de cellules peuvent l’emporter sur plus de profondeur
Une question pratique clé pour tout laboratoire est de savoir s’il faut séquencer chaque cellule très profondément ou profiler davantage de cellules à moindre profondeur. En faisant varier systématiquement le nombre de cellules et le nombre de fragments d’ADN par cellule, les auteurs montrent que la performance des modèles se dégrade très peu lorsque la profondeur de séquençage est réduite à un niveau modéré, à condition qu’un nombre suffisant de cellules soit inclus. En revanche, réduire le nombre de cellules nuit clairement à la précision du modèle, en particulier lorsqu’on évalue la performance sur de nombreux types cellulaires simultanément. Parce que HyDrop v2 est beaucoup moins cher par cellule, les chercheurs peuvent facilement ajouter des dizaines de milliers de cellules supplémentaires, retrouvant ou même dépassant la performance des modèles commerciaux à une fraction du coût.
Voir les empreintes protéiques sur l’ADN
L’étude examine aussi si les différentes plateformes de laboratoire introduisent des biais subtils dans la manière dont l’enzyme ATAC‑seq coupe l’ADN, ce qui pourrait induire en erreur les modèles qui cherchent à déduire où les protéines se trouvent sur le génome. À l’aide d’un outil neuronal distinct qui corrige les préférences de l’enzyme, les auteurs montrent que HyDrop v2 et les kits commerciaux produisent des schémas d’activité enzymatique presque identiques dans les cellules de souris et de drosophile. Après correction, les deux jeux de données révèlent de fines « empreintes » où les protéines régulatrices et les nucléosomes semblent protéger l’ADN de la coupure, et ces empreintes correspondent aux motifs de séquence mis en valeur par les modèles séquence‑vers‑fonction. Cet accord suggère que les plateformes open‑source et commerciales sont également adaptées aux études détaillées des interactions protéines‑ADN.
Pourquoi cela compte pour le décodage du génome
Pour les non‑spécialistes, le message essentiel est que nous pouvons désormais construire des cartes très grandes et abordables de l’utilisation de l’ADN dans les cellules individuelles, et entraîner des modèles d’apprentissage profond puissants sur ces cartes sans dépendre uniquement de matériel propriétaire coûteux. HyDrop v2 fournit des données qui soutiennent la prédiction des enhancers, l’interprétation des motifs de séquence et les empreintes de liaison protéique au même niveau que les méthodes commerciales de pointe, à condition qu’un nombre suffisant de cellules soit profilé. Cela ouvre la porte à la construction d’atlas à l’échelle des organismes des éléments régulateurs en santé et en maladie, accélérant les efforts pour lire les instructions régulatrices du génome et concevoir de nouveaux interrupteurs génétiques ciblés avec précision pour la recherche et de futures thérapies.
Citation: Dickmänken, H., Wojno, M., Mahieu, L. et al. Evaluating single-cell ATAC-seq atlasing technologies using sequence-to-function modeling. Nat Commun 17, 1951 (2026). https://doi.org/10.1038/s41467-026-68742-4
Mots-clés: single‑cell ATAC‑seq, enhancers, modèles d’apprentissage profond, régulation génique, génomique open‑source