Clear Sky Science · fr
Prédiction fiable des numéros de classification des enzymes à l’aide d’un transformeur hiérarchique interprétable
Pourquoi il est important de prédire les fonctions des enzymes
Chaque cellule vivante fonctionne grâce à une multitude de petites machines chimiques appelées enzymes. Chaque enzyme a un « métier » spécifique, codé par un numéro de la Commission des Enzymes (EC), un code en quatre parties un peu comme une adresse postale. L’attribution correcte des numéros EC est cruciale pour comprendre le métabolisme, concevoir de nouveaux médicaments, modifier des microbes afin de produire des carburants ou des alternatives aux plastiques, et suivre la façon dont les écosystèmes transforment les composés chimiques. Mais les expériences pour déterminer la fonction d’une enzyme sont lentes et coûteuses. Cette étude présente HIT-EC, un nouveau modèle d’intelligence artificielle capable de prédire de manière fiable les numéros EC à partir de séquences protéiques tout en expliquant pourquoi il a formulé chaque prédiction.
Un système de code postal pour les fonctions enzymatiques
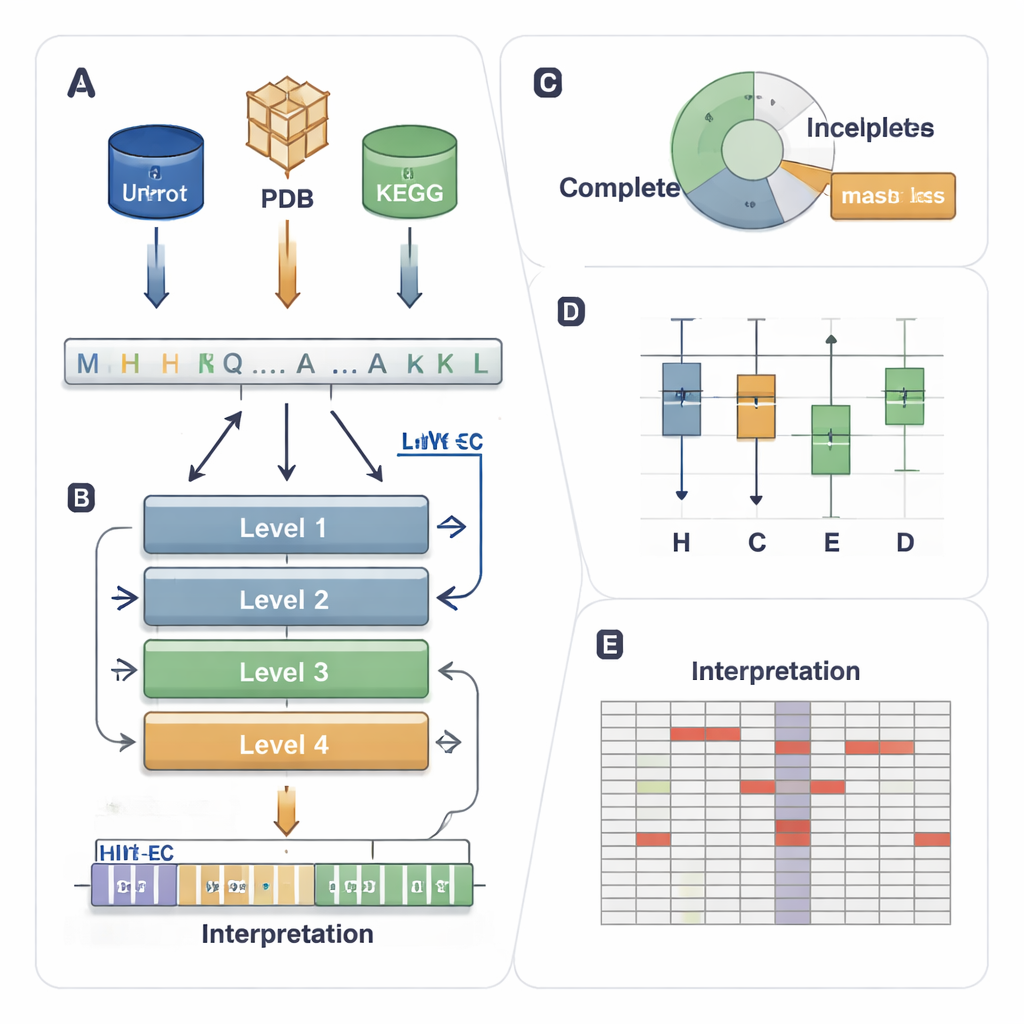
Le système EC attribue à chaque enzyme un code en quatre niveaux tel que 1.1.1.37. Le premier chiffre indique une classe large (par exemple, des enzymes qui déplacent des électrons ou transfèrent des groupes), et les chiffres suivants décrivent des détails réactionnels plus précis. Cette hiérarchie est puissante mais crée un problème de prédiction exigeant : un modèle doit trouver correctement les quatre niveaux parmi des milliers de codes possibles, même lorsque certaines enzymes sont rares ou partiellement annotées dans les bases de données (par exemple 3.5.-.-, où les niveaux détaillés manquent). Les méthodes informatiques existantes utilisent soit la structure 3D, soit la similarité de séquence, soit l’apprentissage profond, mais elles peinent souvent avec les enzymes peu fréquentes, ignorent les données partiellement étiquetées et se comportent généralement comme des « boîtes noires » offrant peu d’explication sur leurs décisions.
Un IA à quatre étages qui suit l’échelle EC
HIT-EC (Hierarchical Interpretable Transformer for EC prediction) est conçu pour refléter la hiérarchie en quatre niveaux du système EC. Il prend une séquence protéique brute et la fait passer par quatre couches de transformeur, chacune axée sur un niveau EC. Des flux locaux relient chaque niveau au précédent, garantissant qu’une décision fine (le quatrième chiffre) soit cohérente avec des décisions plus larges (le premier et le deuxième chiffre). En parallèle, un flux global maintient le contexte complet de la séquence visible à chaque étape. Le modèle peut aussi être entraîné sur des séquences à étiquetage incomplet, en utilisant une « perte masquée » qui ignore simplement les niveaux EC manquants au lieu d’écarter la séquence. Cela permet à HIT-EC d’apprendre à partir de la grande fraction de protéines dans les bases de données annotées de façon partielle.
Meilleures performances que les concurrents en précision et en rapidité
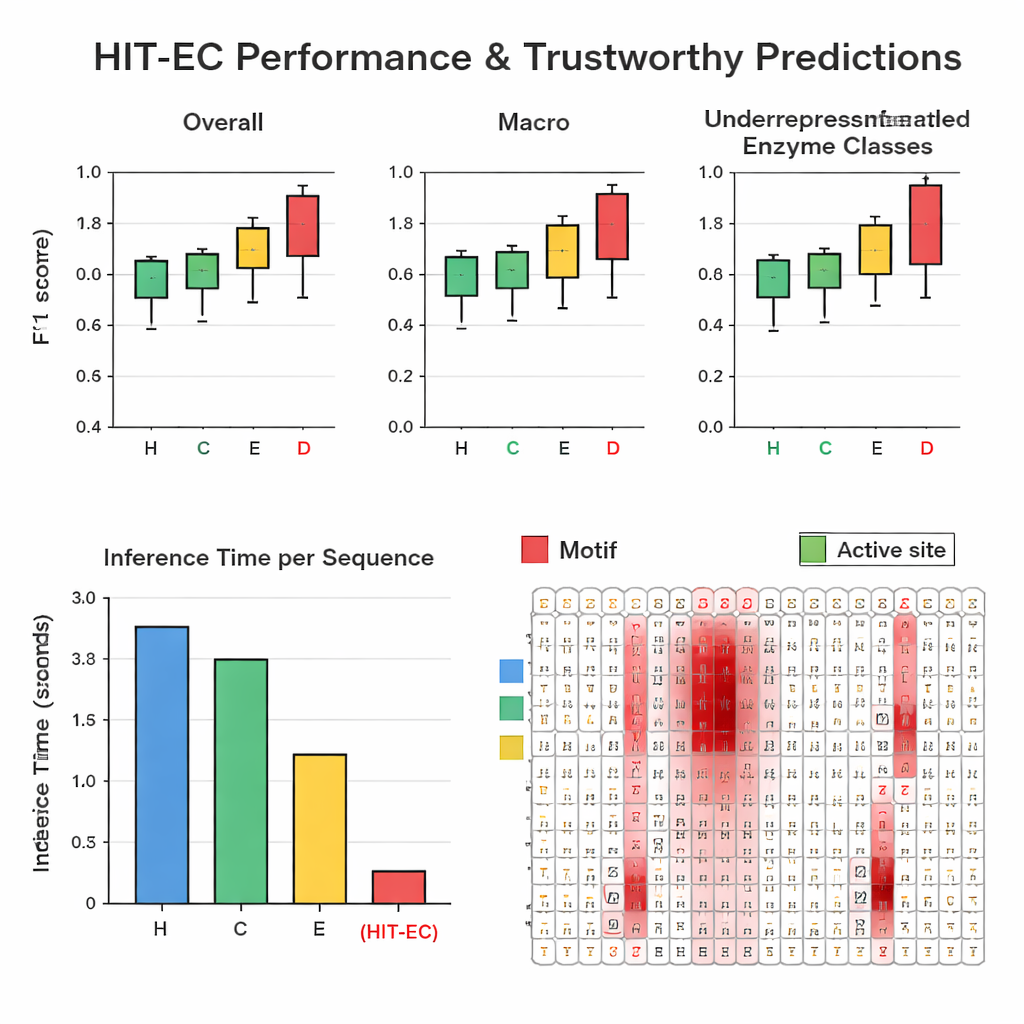
Les auteurs ont constitué un grand jeu de données soigneusement filtré d’environ 200 000 enzymes couvrant 1 938 numéros EC distincts provenant de Swiss-Prot et de la Protein Data Bank. Lors de tests de validation répétés, HIT-EC a surpassé trois méthodes de référence (CLEAN, ECPICK et DeepECtransformer) à la fois sur les scores F1 globaux et par classe, qui mesurent l’équilibre entre vraies positives et fausses alertes. Il s’est montré particulièrement performant sur les codes EC peu représentés (25 exemples ou moins), où les méthodes antérieures échouent souvent. HIT-EC s’est également bien généralisé aux enzymes ajoutées à Swiss-Prot après l’entraînement et à des génomes complets de bactéries diverses, y compris des souches bien étudiées d’Escherichia coli, Bacillus subtilis et Mycobacterium tuberculosis. Malgré sa sophistication, le modèle est très efficace : sur un GPU standard, il a traité une protéine en environ 38 millisecondes — des dizaines de fois plus vite que certains concurrents qui reposent sur des recherches de similarité plus lentes ou des ensembles de modèles nombreux.
Voir ce que le modèle « regarde »
Pour rendre ses prédictions dignes de confiance, HIT-EC est conçu pour montrer quels acides aminés dans la séquence ont influencé chaque décision de niveau EC. Les auteurs ont construit une voie d’interprétation qui combine les poids d’attention avec des informations de gradient pour évaluer l’importance de chaque position. Ils ont validé ces scores sur des familles d’enzymes bien caractérisées. Par exemple, dans une famille de cytochromes P450 (CYP106A2), HIT-EC a mis en évidence des motifs fonctionnels connus tels que les régions de liaison à l’oxygène et à l’hème, et a identifié un motif subtil EXXR que l’un des modèles de référence avait manqué. Pour des représentants classiques de chaque classe EC de premier niveau — comme l’alcool déshydrogénase, l’hexokinase et l’anhydrase carbonique — les scores de pertinence du modèle ont éclairé les motifs signatures et les sites de liaison au substrat décrits dans les manuels. Ces interprétations fournissent des « preuves » biochimiques que le modèle fonde ses prédictions sur des caractéristiques significatives, et non sur des corrélations accidentelles.
Orienter le travail sur les enzymes rares et émergents
L’équipe a testé en outre HIT-EC sur deux enzymes peu étudiées mais importantes pour le nettoyage de la pollution : un cytochrome P450 impliqué dans la dégradation des polluants aromatiques, et une hydrolase dégradant le PET provenant de Streptomyces qui aide à digérer des molécules liées au plastique. Ces deux enzymes avaient été caractérisées expérimentalement mais ne disposaient pas d’attribution EC officielle. HIT-EC a correctement prédit les numéros EC attendus et a mis en évidence des motifs et des résidus catalytiques correspondant aux connaissances issues d’études structurales et biochimiques. Globalement, ce travail montre que HIT-EC peut non seulement attribuer des numéros EC de manière plus précise et plus rapide que les outils actuels, surtout pour des fonctions rares, mais aussi expliquer pourquoi une enzyme donnée est supposée accomplir une tâche chimique particulière. Ce mélange de performance et d’interprétabilité en fait un outil prometteur pour l’annotation fiable et à grande échelle des enzymes en génomique, biotechnologie et recherche environnementale.
Citation: Dumontet, L., Han, SR., Lee, J.H. et al. Trustworthy prediction of enzyme commission numbers using a hierarchical interpretable transformer. Nat Commun 17, 1146 (2026). https://doi.org/10.1038/s41467-026-68727-3
Mots-clés: prédiction de la fonction des enzymes, apprentissage profond en biologie, modèles de transformeur, annotation des protéines, enzymes pour la biorémédiation