Clear Sky Science · fr
Une activation alternative de l’EGFR par la mutation R252C dérivée d’un patient favorise la progression du cancer
Quand les antennes cellulaires se rebellent
Pourquoi certains cancers continuent-ils de progresser malgré des cycles de chimiothérapie et des immunothérapies de pointe ? Cette étude suit un patient présentant des tumeurs au poumon et au cerveau et remonte la maladie à une minuscule modification d’une « antenne » clé à la surface cellulaire nommée EGFR. En dévoilant comment cette seule mutation réoriente les signaux de croissance, les chercheurs expliquent non seulement l’agressivité de la tumeur du patient, mais montrent aussi comment un médicament existant, l’afatinib, peut la contenir.
Une mutation rare aux grandes conséquences
EGFR est un récepteur traversant la membrane cellulaire qui aide les cellules à répondre aux signaux de croissance. De nombreux cancers pulmonaires et cérébraux portent des mutations de l’EGFR, mais la plupart des altérations connues se situent à l’intérieur de la cellule, dans la portion qui fonctionne comme un interrupteur enzymatique. Ici, l’équipe a découvert un changement inhabituel à l’extérieur de l’EGFR, dans la partie qui saisit normalement les facteurs de croissance. Chez un patient atteint à la fois d’un cancer du poumon et d’un gliome, ils ont trouvé qu’un acide aminé à la position 252 avait été remplacé, l’arginine devenant une cystéine — baptisée EGFR R252C. L’exploration des bases de données sur le cancer a montré cette mutation chez une petite fraction de patients atteints de gliome et quasiment jamais dans les tumeurs pulmonaires, suggérant qu’elle est rare mais réelle. À l’aide d’outils d’édition génétique, les auteurs ont recréé cette mutation exacte dans plusieurs lignées cellulaires humaines de cancers du cerveau et du poumon pour tester son effet.
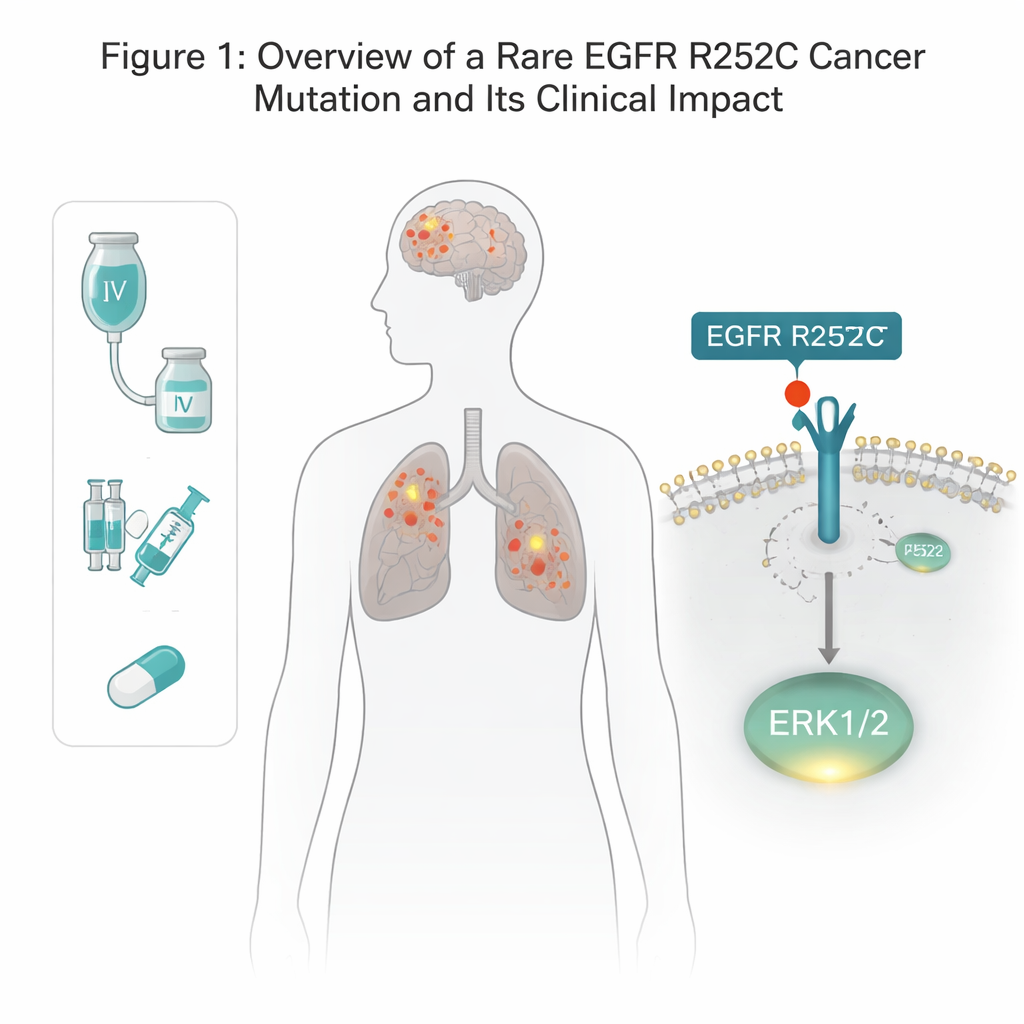
Un nouveau raccourci vers les signaux de croissance
Ordinairement, l’EGFR doit s’apparier avec une copie voisine puis phosphoryler sa propre queue intracellulaire avant d’activer les voies de croissance en aval. Étonnamment, la version R252C n’a pas présenté cette auto-phosphorylation habituelle. À la place, les cellules portant l’EGFR R252C ont activé de façon beaucoup plus marquée un contrôleur de croissance spécifique, ERK1/2, tout en laissant d’autres voies classiques de l’EGFR — comme AKT et STAT3 — largement inchangées. Le blocage d’ERK1/2 avec un inhibiteur dédié a supprimé l’avantage de croissance conféré par R252C, prouvant qu’ERK1/2 est le principal moteur de la puissance tumorale de ce mutant.
Verrouiller le récepteur dans une conformation toujours active
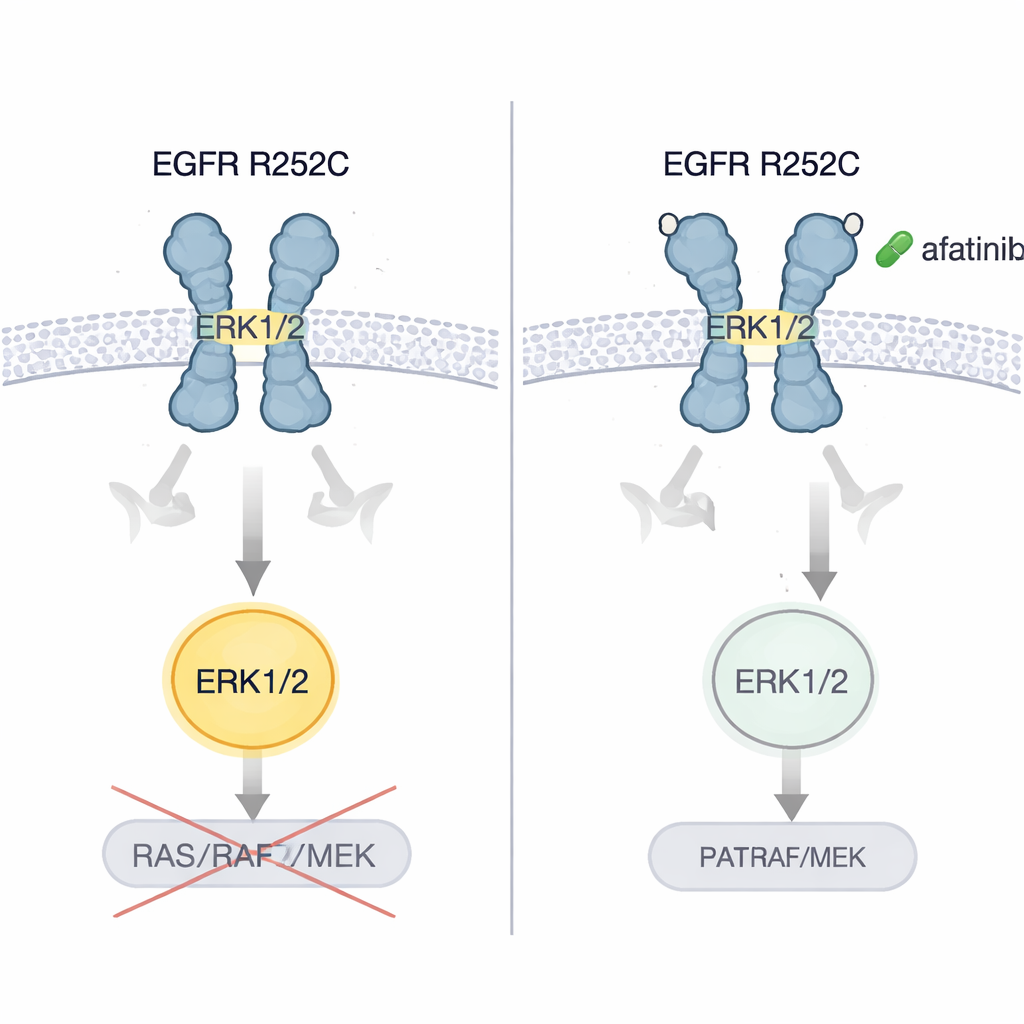
Pour comprendre comment une modification externe pouvait provoquer une telle surrégulation sélective, les chercheurs ont combiné des essais biochimiques et des simulations informatiques. L’échange R252C introduit une nouvelle cystéine à l’extérieur de l’EGFR. Deux mutants de ce type peuvent former une liaison disulfure — une sorte d’agrafage moléculaire — entre leurs résidus C252, les verrouillant ensemble en une paire stable. La modélisation structurelle a montré que cette liaison force l’extérieur du récepteur dans un alignement « en V » décalé qui imite étroitement la forme active liée au ligand, même en l’absence de facteur de croissance. Cet alignement se propage à travers les segments transmembranaires et juxtamembranaires, torsadant les domaines enzymatiques internes dans une configuration inhabituelle : les sites actifs sont orientés vers l’intérieur de la cellule mais sont maintenus trop éloignés pour se phosphoryler efficacement mutuellement. Au lieu de cela, cette conformation crée une forte surface d’ancrage pour ERK1/2, permettant à l’EGFR R252C de phosphoryler directement ERK1/2 et de contourner la relais classique RAS–RAF–MEK.
Des modèles murins à un seul patient
Les auteurs ont montré que des cellules de cancers cérébraux et pulmonaires portant l’EGFR R252C proliféraient plus vite en culture et formaient des tumeurs plus grandes et plus agressives lorsqu’elles étaient implantées chez la souris, comparées à des cellules porteuses d’un EGFR normal. Ils ont ensuite testé plusieurs générations de pilules bloquant l’EGFR. Seul l’afatinib, un inhibiteur de deuxième génération, a systématiquement éteint l’activation d’ERK1/2 et réduit fortement la croissance des cellules tumorales. Dans des modèles murins de tumeurs cérébrales et pulmonaires induites par R252C, l’afatinib a ralenti l’expansion tumorale et prolongé la survie. Fait crucial, lorsque le patient initial — dont la maladie s’était aggravée malgré la chimiothérapie, un médicament ciblant les vaisseaux sanguins et l’immunothérapie — a été mis sous afatinib, les bilans d’imagerie du poumon et du cerveau ont montré une réduction marquée de la charge tumorale et le patient a bénéficié de plusieurs années sans progression.
Quelles implications pour les patients
Ce travail révèle un mode d’action auparavant non reconnu d’une mutation cancérogène de l’EGFR : en agrafant deux récepteurs ensemble à l’extérieur de la cellule, elle les tord dans une pose active qui déclenche directement ERK1/2 au lieu de suivre la chaîne de signalisation classique. Pour un public non spécialiste, l’idée principale est que toutes les mutations d’un même gène ne se comportent pas de la même façon, et que certaines altérations rares peuvent répondre mieux à des médicaments spécifiques existants. L’EGFR R252C semble engendrer des cancers particulièrement vulnérables à l’afatinib. Bien que cette conclusion repose actuellement sur un cas clinique détaillé et des travaux de laboratoire étendus, elle plaide en faveur d’un dépistage plus personnalisé des mutations du domaine externe de l’EGFR et suggère que des thérapies ciblées bien choisies pourraient offrir un nouvel espoir à certains patients atteints de tumeurs cérébrales et pulmonaires difficiles à traiter.
Citation: Zhang, Y., Fei, Q., Li, Y. et al. An alternative EGFR activation by patient-derived R252C mutation promotes cancer progression. Nat Commun 17, 1902 (2026). https://doi.org/10.1038/s41467-026-68699-4
Mots-clés: mutation EGFR, gliome, cancer du poumon, signalisation ERK, afatinib