Clear Sky Science · fr
Améliorer la prédiction par score polygénique pour les groupes sous-représentés par l’apprentissage par transfert
Pourquoi votre score de risque génétique peut ne pas fonctionner pour vous
Les « scores » de risque génétique sont de plus en plus utilisés pour estimer la probabilité qu’une personne développe des maladies courantes comme le diabète, les maladies cardiaques ou l’hypertension. Mais la plupart de ces scores ont été construits à partir de données ADN de personnes d’ascendance européenne. En conséquence, ils prédisent souvent mal pour des personnes provenant d’autres origines, ce qui soulève des questions d’équité et d’utilité en médecine réelle. Cette étude pose une question simple : peut‑on réutiliser ce que nous avons appris à partir de larges jeux de données européens pour construire des scores génétiques meilleurs et plus justes pour des groupes sous‑représentés — sans partager les données brutes de qui que ce soit ?
Des millions de marqueurs ADN à un unique score de risque
Un score polygénique ressemble à une fiche de notes qui additionne les petits effets de nombreux marqueurs génétiques répartis sur le génome. Chaque marqueur reçoit un poids qui reflète la force de son association avec un trait, d’après de grandes études génétiques. Quand ces études concernent majoritairement des Européens, le score obtenu a tendance à mieux fonctionner pour les Européens. Les différences de background génétique — la fréquence de certains variants et la façon dont ils sont hérités ensemble — font que les mêmes poids donnent souvent de mauvais résultats chez les Afro‑Américains, les Hispaniques et d’autres populations. Constituer des jeux de données aussi vastes pour chaque groupe est coûteux et lent, aussi les auteurs se sont tournés vers une stratégie d’apprentissage automatique appelée apprentissage par transfert : au lieu de repartir de zéro pour chaque population, ils affinent un modèle existant entraîné ailleurs.
Comment emprunter des connaissances sans partager les données brutes
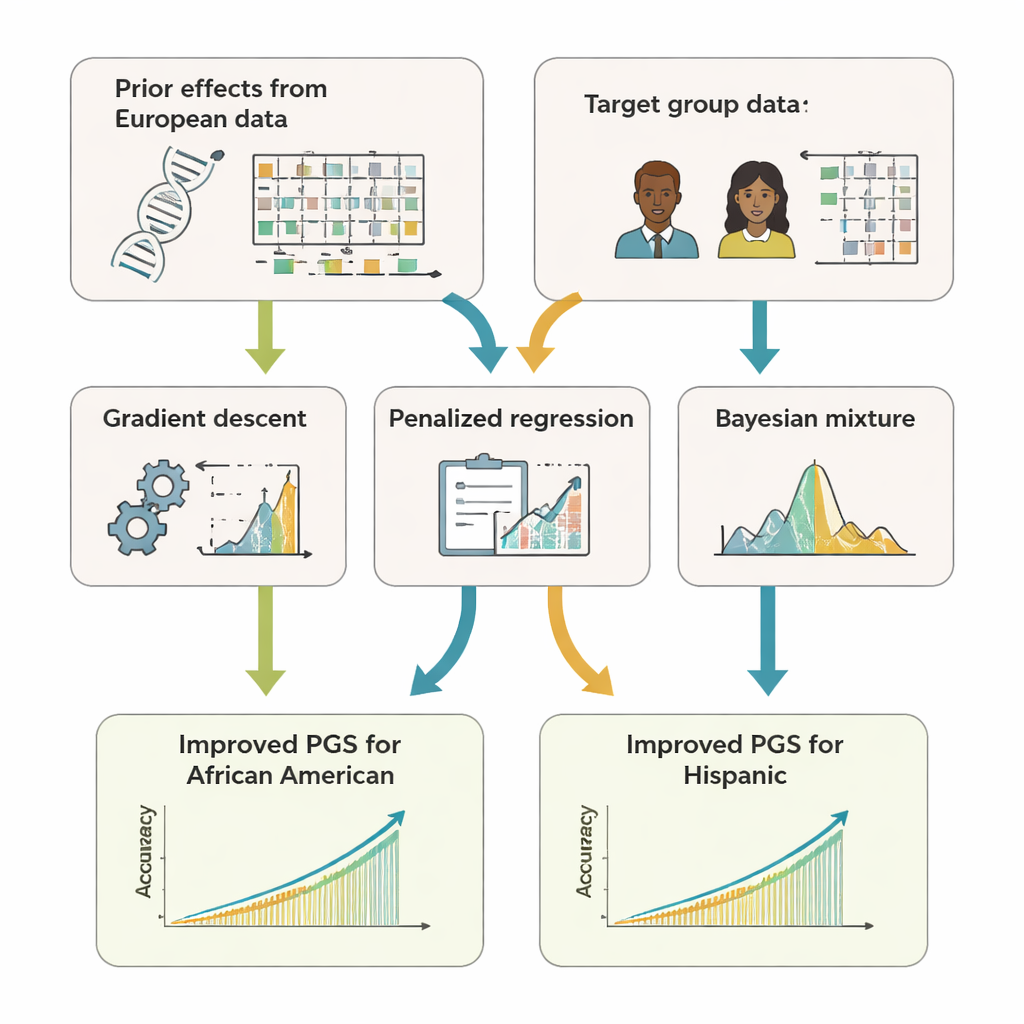
L’équipe a développé GPTL, un paquet logiciel R open source qui implémente trois approches d’apprentissage par transfert pour les scores génétiques. Les trois méthodes partent d’estimations existantes des effets des variants obtenues dans un large jeu de données d’ascendance européenne, puis ajustent progressivement ces estimations à l’aide de données d’un groupe cible, comme des Afro‑Américains ou des Hispaniques. Une méthode modifie les poids européens pas à pas avec une descente de gradient et s’arrête tôt, avant de les remplacer complètement. Une seconde méthode, la régression pénalisée, attire activement les nouvelles estimations vers les valeurs originales à moins que les données cibles n’apportent une preuve solide du contraire. La troisième, un modèle de mélange bayésien, permet à chaque marqueur ADN de choisir parmi plusieurs sources d’information — comme plusieurs groupes d’ascendance ou même une option « aucun effet » — et les combine selon leur capacité à expliquer les données cibles.
Mettre les méthodes à l’épreuve
Pour évaluer ces approches, les auteurs ont utilisé à la fois des simulations informatiques et des données réelles provenant de centaines de milliers de volontaires du UK Biobank et du programme U.S. All of Us. Ils se sont concentrés sur des participants afro‑américains et hispaniques comme groupes cibles et ont utilisé des données d’ascendance européenne comme source principale d’information a priori. Sur 11 traits — notamment la taille, l’indice de masse corporelle, les lipides sanguins, la pression artérielle et des marqueurs rénaux — les scores obtenus par apprentissage par transfert ont systématiquement mieux prédit que les scores construits uniquement à l’intérieur du groupe cible ou réutilisés tels quels depuis les Européens. Souvent, leur précision égalait ou dépassait légèrement celle de méthodes « multi‑ascendance » plus complexes qui nécessitent de combiner des données brutes de plusieurs populations. De manière cruciale, les méthodes de GPTL n’ont besoin que de statistiques sommaires — des chiffres agrégés sur les effets génétiques — ce qui permet à des institutions de collaborer sans exposer les données génétiques individuelles.
Quand plus d’ADN n’est pas toujours mieux
Les chercheurs ont aussi examiné comment choisir au mieux les marqueurs génétiques à inclure. Contrairement à l’idée répandue selon laquelle utiliser tous les marqueurs disponibles aide toujours, ils ont trouvé que pour les groupes afro‑américains et surtout hispaniques, inclure des millions de signaux très faibles pouvait en fait nuire à la performance, en particulier lorsqu’on utilisait des représentations très simplifiées des corrélations génétiques. Se concentrer sur des marqueurs mieux étayés et utiliser des informations plus riches sur la façon dont les variants sont hérités ensemble aboutissait souvent à des scores plus précis. L’étude montre également que l’ajout d’informations a priori provenant de plusieurs groupes d’ascendance et une modélisation soigneuse des différences entre populations amélioraient encore les prédictions.
Ce que cela signifie pour une prédiction génétique plus équitable
Pour les populations non européennes, les scores de risque génétique disponibles aujourd’hui peuvent être nettement moins performants, ce qui risque d’aggraver les inégalités en santé. Ce travail démontre que l’apprentissage par transfert — affiner intelligemment des scores basés sur des données européennes avec des jeux de données modestes issus de groupes sous‑représentés — peut réduire une grande partie de cet écart. Concrètement, cela signifie que les systèmes de santé et les chercheurs peuvent construire des outils génétiques plus précis et plus équitables sans fusionner les données brutes entre institutions ou entre ascances, ce qui atténue les préoccupations relatives à la vie privée. Aucun méthode unique ne sera optimale pour tous les traits et toutes les populations, mais la boîte à outils GPTL montre que la prédiction génétique plus juste est techniquement à portée de main si l’on considère les modèles passés non comme des produits figés, mais comme des points de départ adaptables à tous.
Citation: Wu, H., Pérez-Rodríguez, P., Boehnke, M. et al. Improving polygenic score prediction for underrepresented groups through transfer learning. Nat Commun 17, 1973 (2026). https://doi.org/10.1038/s41467-026-68696-7
Mots-clés: scores de risque polygénique, apprentissage par transfert, prédiction génétique, inégalités en santé, génétique des populations