Clear Sky Science · fr
ProteoAutoNet : analyse haut débit de protéines co-élutées avec robotique et apprentissage automatique
Pourquoi comprendre les partenariats protéiques est important
À l’intérieur de chaque cellule, les protéines travaillent rarement seules. Elles s’associent dans des alliances changeantes pour construire des structures, copier l’ADN, éliminer des éléments endommagés et fournir l’énergie de la croissance. De nombreux cancers détournent ces partenariats, mais les cartographier en détail a été un travail lent et minutieux. Cette étude présente ProteoAutoNet, un système reposant sur la robotique et l’apprentissage automatique qui accélère considérablement la découverte des partenariats protéiques dans les cellules, et montre comment cette approche peut révéler des points faibles cachés dans les cancers de la thyroïde.
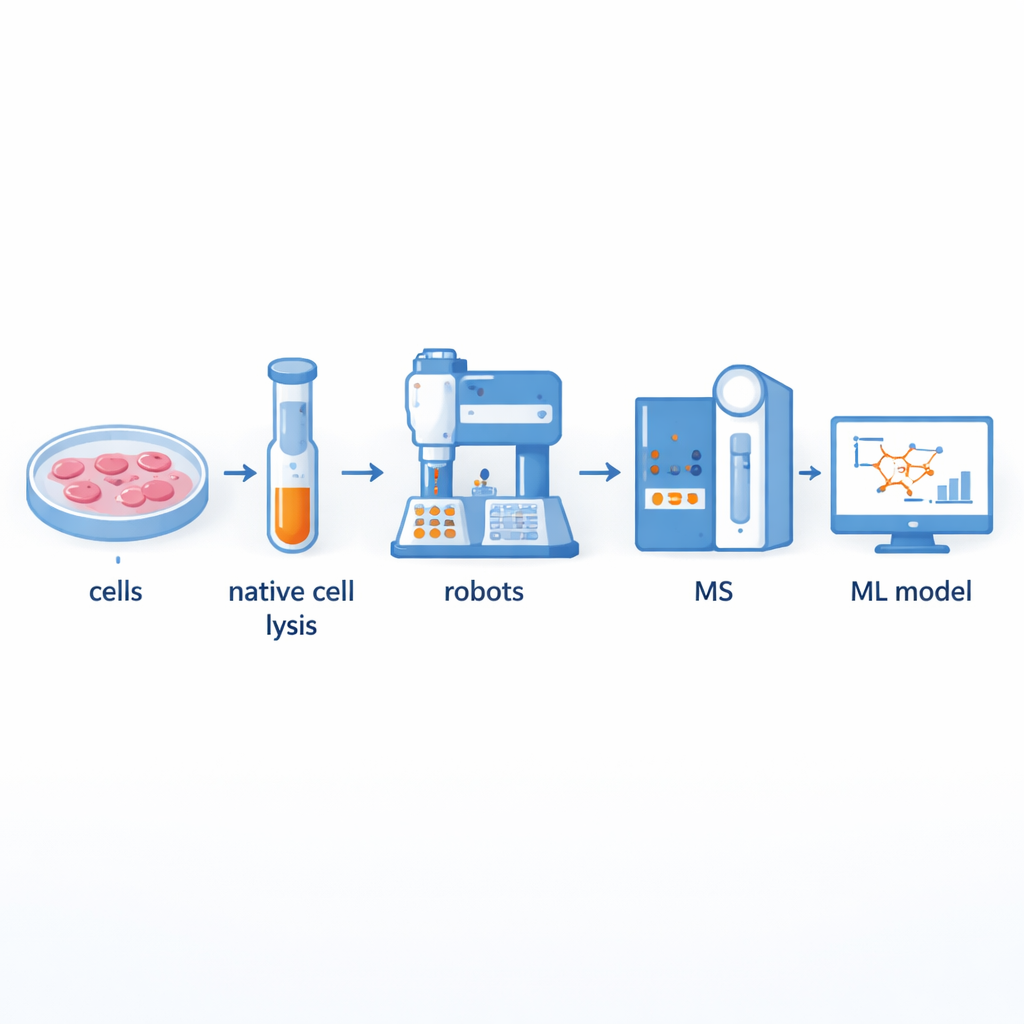
Construire une usine de partenariats protéiques plus rapide
Traditionnellement, les chercheurs utilisent une méthode nommée spectrométrie de masse après co-fractionnement pour séparer les grands complexes protéiques puis identifier leurs composants. Bien que puissante, cette approche est gourmande en main‑d’œuvre et à faible débit : préparer des centaines de fractions à la main peut prendre plusieurs jours. Les auteurs ont développé une plateforme assistée par robotique qui automatise la majeure partie de ce flux de travail. Le contenu cellulaire est d’abord délicatement découpé pour préserver les complexes protéiques naturels, puis passé à travers des colonnes de séparation selon la taille pour être divisé en dizaines de fractions. Des robots de manipulation de liquides et des bras robotiques prennent ensuite le relais, ajoutant des réactifs, digérant les protéines en fragments, nettoyant les échantillons et les acheminant vers un spectromètre de masse pour mesure. Cet agencement peut traiter jusqu’à 540 fractions issues de plusieurs lignées cellulaires thyroïdiennes en seulement deux à trois jours, doublant environ le débit comparé aux systèmes semi-automatisés précédents.
Des robots non seulement plus rapides, mais aussi plus fiables
La vitesse seule ne suffit pas si les résultats sont bruyants ou incohérents. L’équipe a vérifié attentivement si la chaîne robotisée égalait ou dépassait la qualité du traitement manuel traditionnel. À l’aide d’échantillons de contrôle qualité, ils ont montré que le système automatisé identifiait régulièrement près de 3 000 protéines par lignée cellulaire thyroïdienne avec un fort recouvrement entre réplicats et une grande concordance des quantités mesurées. Lors d’une comparaison directe entre traitement robotisé et manuel des mêmes échantillons, les deux approches détectaient des nombres de protéines similaires, mais la méthode robotisée présentait une variation légèrement moindre dans les comptages et des mesures d’abondance protéique plus stables. Cela signifie que la nouvelle plateforme permet non seulement d’économiser du temps et du travail, mais aussi de mener des expériences plus reproductibles — une exigence cruciale pour les grandes études et les applications cliniques.
Apprendre aux ordinateurs à reconnaître les connexions significatives
Même avec des instruments rapides, un défi central demeure : décider quelles protéines interagissent réellement et lesquelles se retrouvent ensemble par hasard. Pour y répondre, les auteurs ont combiné des bases de données de complexes protéiques expertisées avec un modèle d’apprentissage automatique basé sur l’algorithme XGBoost. Ils ont d’abord nettoyé et fusionné trois ressources majeures de complexes protéiques, obtenant 96 635 interactions protéine–protéine connues. Ils ont ensuite utilisé les profils d’apparition des protéines à travers les fractions comme caractéristiques d’entrée, et étiqueté les paires comme partenaires probables ou non en se basant sur les bases de données. Parce que les partenariats réels et de haute confiance sont relativement rares, ils ont utilisé une stratégie ciblée d’augmentation des données : ils ont généré de nombreuses versions légèrement perturbées d’exemples positifs connus pour apprendre au modèle à reconnaître des motifs robustes plutôt qu’à mémoriser des traces spécifiques. Entraîné sur des dizaines de millions d’exemples de ce type issus de trois lignées thyroïdiennes, le modèle a obtenu de bonnes performances, en classant correctement les vraies interactions bien au‑dessus du hasard tant dans des tests internes que dans une lignée de validation indépendante.
Nouvelles perspectives sur la machinerie des cellules cancéreuses
Armés de ce flux de travail, les chercheurs ont cartographié les réseaux d’interaction dans une lignée cellulaire thyroïdienne normale et deux lignées cancéreuses : une carcinome papillaire et une carcinome folliculaire capable de métastaser aux poumons. Dans ces cellules, ils ont identifié plus de 25 000 interactions protéiques probables et mis en évidence des signaux forts provenant de machines cellulaires bien connues telles que les ribosomes (qui synthétisent les protéines) et les protéasomes (qui les dégradent), confirmant que la méthode retrouve la biologie établie. En comparant les cancers à la lignée normale, ils ont mis au jour des réseaux dont l’activité était augmentée en maladie. Dans les cellules de carcinome folliculaire métastatique, à la fois les composants du protéasome et un complexe chaperon nommé préfoldine étaient sensiblement plus connectés et abondants. Plusieurs sous‑unités de préfoldine avaient déjà été liées à d’autres cancers, mais les enquêtes protéiques globales avaient manqué leur comportement coordonné dans le cancer thyroïdien, peut‑être parce que ces protéines sont strictement régulées par la dégradation. L’approche par co-fractionnement a permis de révéler leurs changements coordonnés au niveau des complexes.
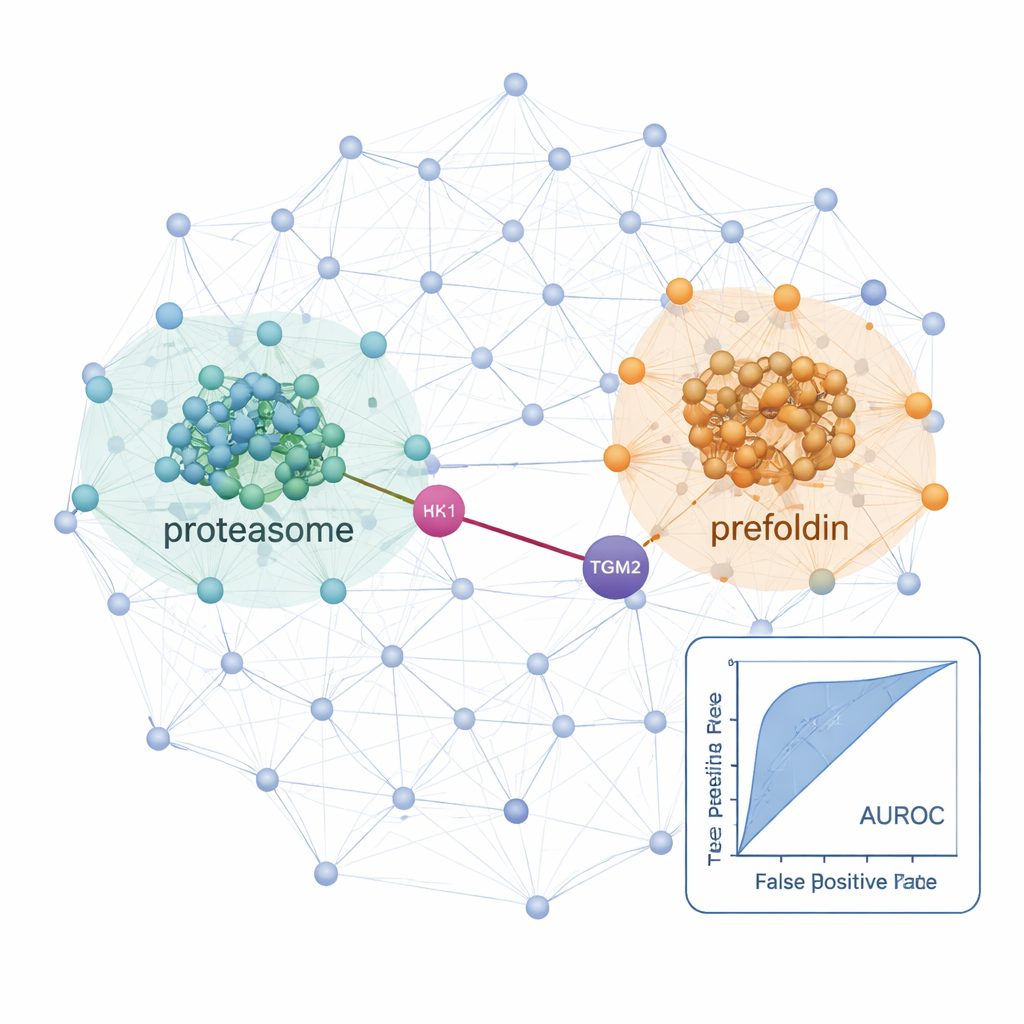
Liens cachés qui pourraient orienter des traitements futurs
L’étude a également mis en lumière des interactions spécifiques susceptibles d’influencer la croissance et la dissémination des cancers thyroïdiens. Un exemple est un partenariat prédit entre HK1, une enzyme qui initie la principale voie de combustion du glucose de la cellule, et TGM2, une protéine connue pour favoriser l’invasion et les métastases dans les tumeurs thyroïdiennes. Cette connexion HK1–TGM2, absente des bases de données d’interactions existantes, a été soutenue par une modélisation structurale et semblait particulièrement active dans la lignée de carcinome papillaire, suggérant que la reprogrammation métabolique et le comportement invasif pourraient être reliés physiquement. En somme, ProteoAutoNet montre comment la combinaison de la robotique et de l’apprentissage automatique peut transformer la cartographie des réseaux protéiques, jusque‑là lente et réservée aux experts, en un processus plus évolutif. Pour les non‑spécialistes, le message clé est que cette technologie peut mettre au jour à la fois des modifications globales de la machinerie cellulaire et des partenariats protéiques inattendus qui pourraient un jour aider les médecins à mieux prédire quels cancers thyroïdiens seront agressifs et à proposer de nouvelles cibles thérapeutiques.
Citation: Lyu, M., Hu, P., Zhang, G. et al. ProteoAutoNet: high-throughput co-eluted protein analysis with robotics and machine learning. Nat Commun 17, 1949 (2026). https://doi.org/10.1038/s41467-026-68686-9
Mots-clés: interactions protéiques, spectrométrie de masse, apprentissage automatique en biologie, cancer de la thyroïde, protéasome et préfoldine