Clear Sky Science · fr
Évaluation complète des méthodes de score polygénique mono et multi‑ancestries avec la plateforme PGS‑hub
Pourquoi votre score de risque génétique compte
Les médecins deviennent meilleurs pour lire notre ADN et estimer qui est plus susceptible de développer des maladies courantes comme les maladies cardiovasculaires, le diabète ou la schizophrénie. Ces estimations, appelées scores polygéniques, combinent les effets minimes de nombreux variants génétiques en un seul chiffre. Mais il existe aujourd’hui de nombreuses méthodes concurrentes pour calculer ces scores, et elles ne fonctionnent pas de manière équivalente pour des personnes d’origines ancestrales différentes. Cette étude visait à comparer les méthodes phares entre elles et à construire un service en ligne, PGS‑hub, qui permet aux chercheurs de calculer ces scores de façon cohérente et simple.
Une plateforme tout-en-un pour calculer les risques ADN
Les auteurs ont créé PGS‑hub, une plateforme web qui masque une grande partie de la complexité technique des scores polygéniques. Les utilisateurs téléversent les résultats d’études génétiques résumant comment des millions de marqueurs d’ADN se rapportent à une maladie ou un trait. Ils choisissent ensuite l’origine ancestrale de la population qui les intéresse — par exemple européenne ou africaine — et sélectionnent dans un menu les méthodes de score populaires. En coulisses, PGS‑hub convertit les entrées aux formats requis, branche des panneaux de référence préconstruits décrivant la corrélation entre marqueurs voisins, et exécute un grand nombre de tâches sur un système de calcul haute performance. Le résultat est un fichier compact de poids pouvant être appliqué à des génomes individuels pour générer un score pour chaque personne. 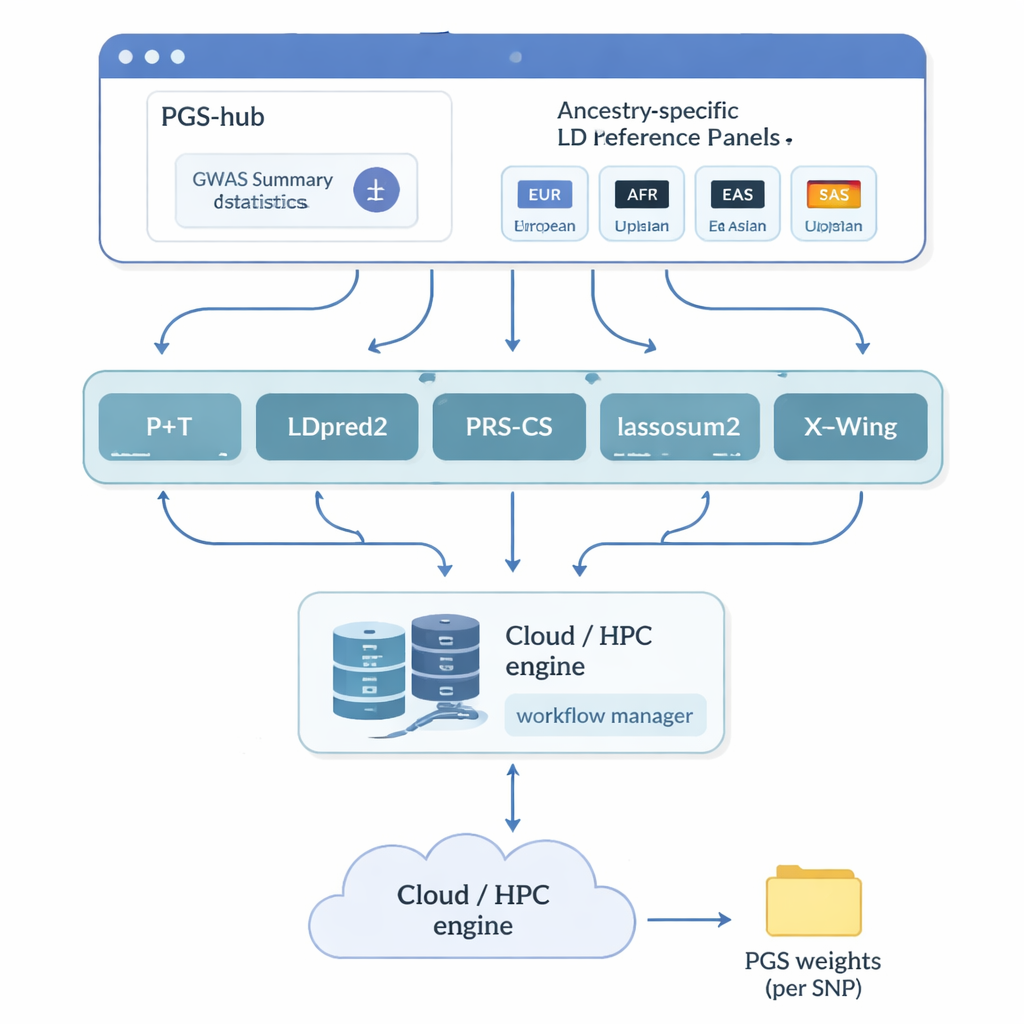
Mise à l’épreuve de 13 méthodes de score
Pour déterminer quelles approches fonctionnent le mieux, l’équipe a comparé 13 méthodes à la pointe sur 36 maladies et traits chez près de 380 000 personnes d’ascendance européenne et un peu plus de 8 000 personnes d’ascendance africaine issues de l’UK Biobank. Ils ont évalué non seulement la capacité de chaque score à prédire qui avait une maladie ou une valeur de trait plus élevée, mais aussi le temps de calcul et la mémoire consommés par chaque méthode. Chez les Européens, une méthode appelée LDpred2 a généralement fourni les scores les plus précis, devançant souvent les autres de manière nette. Quelques alternatives — lassosum2, PRS‑CS et SDPR — ont obtenu des performances proches pour de nombreux traits, tandis que certaines méthodes plus anciennes restaient à la traîne. Pour des traits comme la taille ou la maladie de Crohn, les meilleurs scores expliquaient une part substantielle du risque génétique ; pour d’autres, comme la fonction rénale, toutes les méthodes ont peiné, reflétant des signaux génétiques sous‑jacents plus faibles.
Enseignements pour des populations diverses et les méthodes combinées
Un enjeu majeur en prédiction génétique est que des méthodes entraînées principalement sur des Européens peuvent mal se transférer à des personnes d’origines différentes. Lorsque les auteurs ont répété leurs benchmarks en utilisant des études génétiques d’ascendance africaine, chaque méthode a obtenu des performances plus faibles, mettant en évidence le manque de grandes cohortes dans ces populations. Néanmoins, LDpred2 et SDPR figuraient souvent parmi les meilleures options. L’équipe a aussi examiné des approches « multi‑ancestries » qui combinent explicitement l’information entre populations. Ici, une stratégie relativement simple — combiner linéairement les meilleurs scores LDpred2 spécifiques à chaque ancestralité en un seul score LDpred2‑multi — a surpassé des modèles multi‑ancestries plus élaborés tels que PRS‑CSx et X‑Wing pour les groupes européen et africain. De plus, les auteurs ont montré que construire un ensemble, qui mélange les scores les plus performants issus de plusieurs méthodes, améliorait encore la prédiction pour l’ensemble des traits, en particulier pour des maladies fortement héritables comme la schizophrénie et la cardiopathie coronarienne. 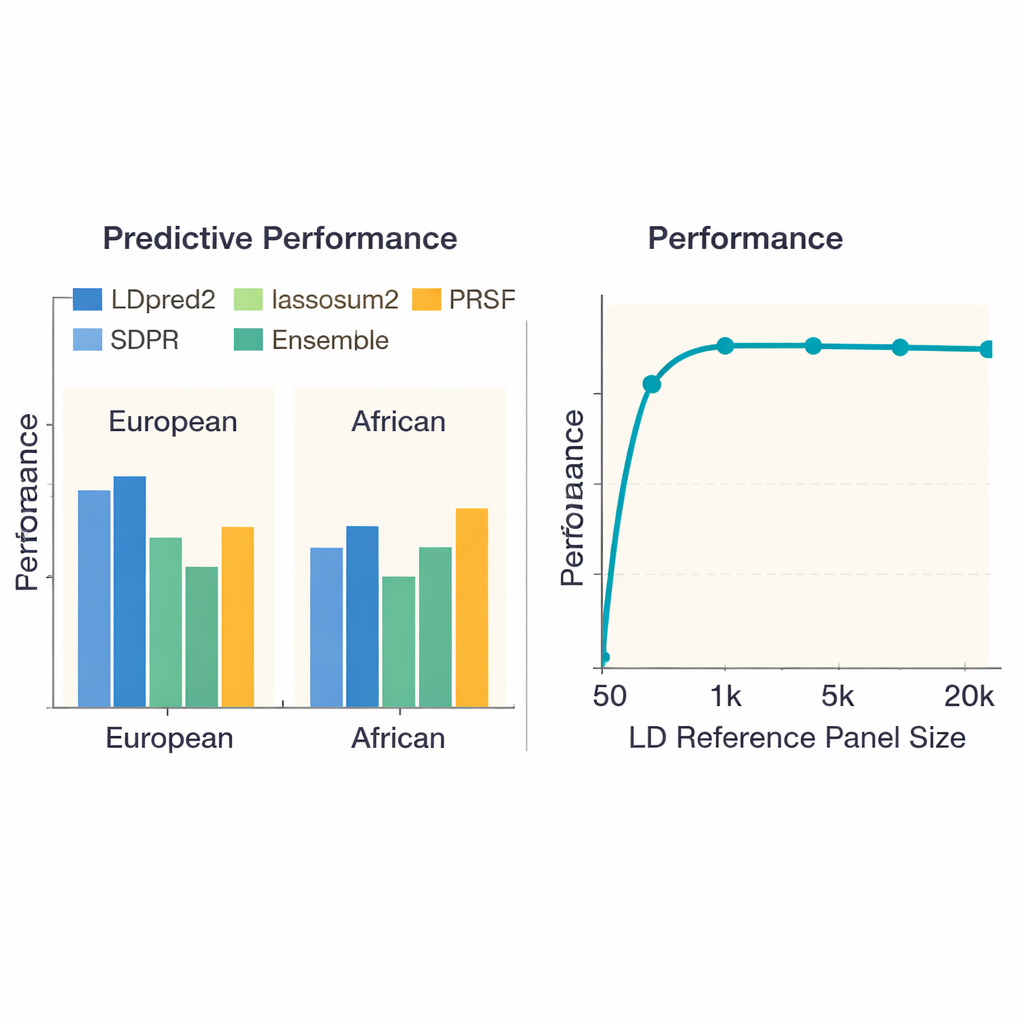
Comment les choix de données et les limites informatiques influent sur les scores
L’étude a sondé l’impact de la taille du panneau de référence — l’ensemble de personnes utilisé pour apprendre comment les marqueurs d’ADN voisins covarient — sur les performances. Lorsque ce panneau était très réduit (moins de 1 000 individus), les scores étaient sensiblement moins précis. À mesure que le panneau atteignait environ 5 000 personnes, les performances s’amélioraient fortement puis se stabilisaient, suggérant que des panneaux de plus en plus grands apportent des gains marginaux décroissants. De façon surprenante, ajouter simplement plus de marqueurs d’ADN n’a pas toujours aidé : l’utilisation d’environ 6,6 millions de variants a parfois détérioré les prédictions par rapport à un jeu soigneusement choisi d’environ 1,1 million, probablement parce que les variants supplémentaires ajoutaient plus de bruit que de signal utile. Les auteurs ont aussi documenté de grandes différences de coût informatique. Des méthodes simples comme le pruning‑and‑thresholding basique se terminaient en moins d’une heure par trait, tandis que certaines approches bayésiennes exigeaient des centaines d’heures CPU — une information importante pour les projets de grande envergure ou les équipes aux ressources limitées.
Ce que cela signifie pour la prédiction génétique future
Pour un public non spécialiste, le message principal est que tous les scores de risque génétique ne se valent pas, et que les détails de leur construction influencent fortement qui en bénéficiera. Ce travail fournit des recommandations pratiques : des méthodes comme LDpred2 et des ensembles bien conçus ont tendance à fournir les prédictions les plus fiables dans de larges jeux de données européens, et les combinaisons multi‑ancestries peuvent surpasser des modèles trans‑population plus complexes. Dans le même temps, la baisse de précision chez les individus d’ascendance africaine souligne le besoin urgent de cohortes génétiques plus grandes et plus diverses. En regroupant de nombreuses méthodes dans une plateforme en ligne standardisée, PGS‑hub abaisse la barrière pour que des chercheurs du monde entier génèrent et comparent des scores polygéniques, une étape importante pour utiliser ces scores de manière équitable et efficace en médecine.
Citation: Chen, X., Wang, F., Zhao, H. et al. Comprehensive benchmarking single and multi ancestry polygenic score methods with the PGS-hub platform. Nat Commun 17, 2014 (2026). https://doi.org/10.1038/s41467-026-68599-7
Mots-clés: scores polygéniques, prévision du risque génétique, plateforme PGS‑hub, génomique multi‑ancestrale, UK Biobank