Clear Sky Science · fr
TET1 en tant que régulateur maître contrôlant la surveillance de la ferroptose dépendante et indépendante de GPX4 dans la leucémie myéloïde aiguë
Pourquoi cette recherche compte pour le traitement du cancer
De nombreux nouveaux médicaments anticancéreux cherchent à pousser les cellules malignes vers un mode d’autodestruction appelé ferroptose, un type de mort cellulaire lié au fer et aux dommages lipidiques. Pourtant, certaines tumeurs résistent obstinément à cette approche. Cette étude révèle comment une protéine modifiant l’ADN nommée TET1 aide les cellules leucémiques à échapper à la ferroptose via deux systèmes de défense biochimiques distincts — et montre que bloquer ces défenses peut rendre vulnérables même des cancers résistants.
Un mélange mortel de fer et de lipides endommagés
La ferroptose survient lorsque le fer alimente l’oxydation incontrôlée des lipides des membranes cellulaires, provoquant finalement la rupture des cellules. Dans la leucémie myéloïde aiguë (LMA), comme dans de nombreux cancers, les cellules déploient de puissants systèmes de surveillance pour tenir ce processus en échec. Un gardien clé est l’enzyme GPX4, qui utilise une petite molécule appelée glutathion pour neutraliser les hydroperoxydes lipidiques nocifs. D’autres systèmes de secours génèrent des molécules antioxydantes capables de piéger des radicaux dangereux même lorsque GPX4 est compromis. Comprendre quels interrupteurs maîtres coordonnent ces défenses est crucial pour concevoir des thérapies qui déclenchent de manière fiable la ferroptose dans les cellules cancéreuses tout en préservant les tissus sains.
TET1 émerge comme un centre de contrôle central
Les chercheurs ont comparé des dizaines d’échantillons de cellules cancéreuses, y compris de nombreuses lignées de LMA et des cellules dérivées de patients, et ont remarqué un schéma clair : les cellules résistantes à la ferroptose présentaient des niveaux plus élevés de TET1, une enzyme qui modifie les marques chimiques de l’ADN et influence l’activité des gènes. Lorsqu’ils ont réduit les niveaux de TET1 à l’aide d’outils génétiques ou inhibé son activité avec une petite molécule, les cellules cancéreuses sont devenues nettement plus sensibles aux médicaments induisant la ferroptose. Cela s’est vérifié tant en culture qu’en modèles murins de LMA. Inversement, augmenter l’expression de TET1 protégeait les cellules de la mort ferroptotique et limitait l’accumulation d’espèces réactives de l’oxygène, ces sous-produits chimiquement agressifs qui provoquent les dommages membranaires.
Renforcer le bouclier antioxydant principal
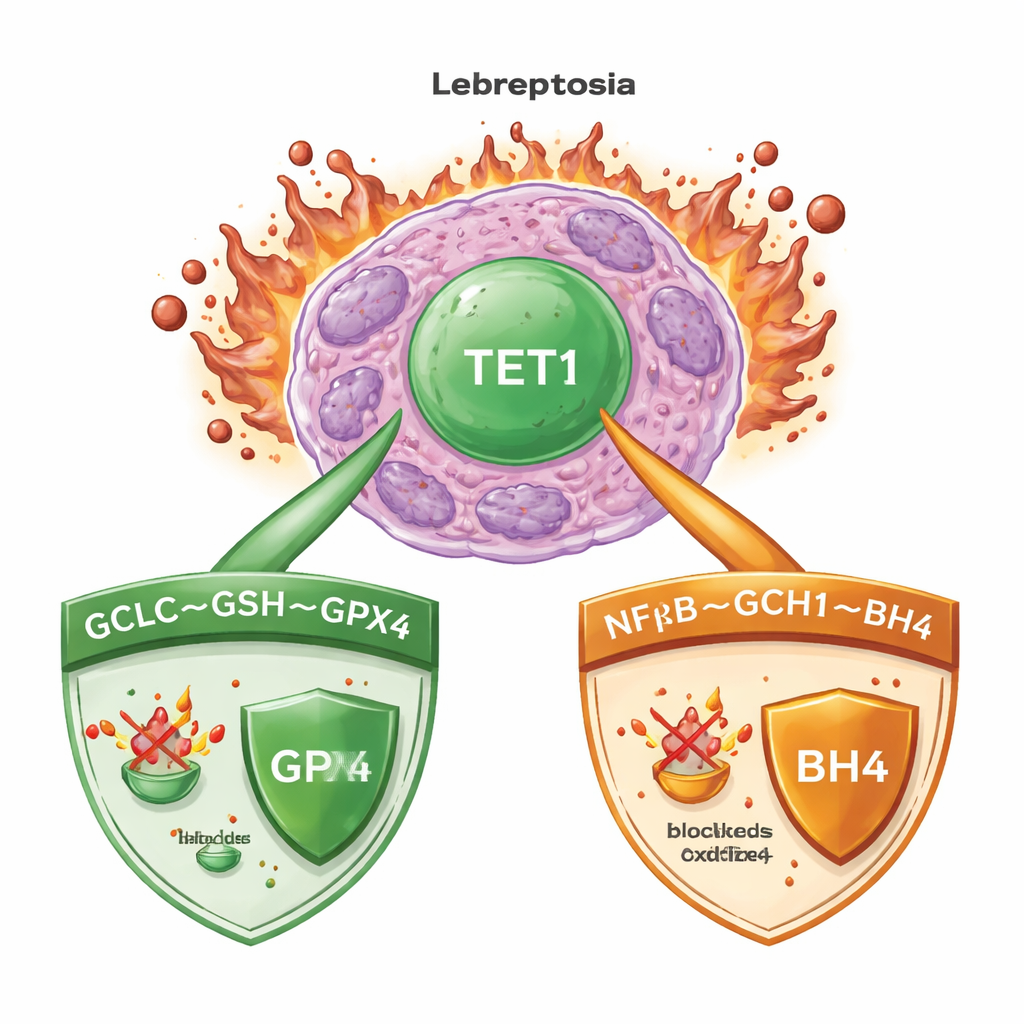
En creusant plus avant, l’équipe a cartographié les sites d’action de TET1 sur le génome et a constaté qu’il active directement un gène appelé GCLC. GCLC code pour une enzyme cruciale qui lance la production de glutathion, le carburant de GPX4. En augmentant une marque d’ADN spécifique (5-hydroxyméthylcytosine) au niveau du promoteur de GCLC, TET1 stimule la synthèse de glutathion. Dans des conditions nutritionnelles normales, cela renforce le pool antioxydant principal de la cellule. En cas de privation en cystine, le même complexe enzymatique fabrique aussi des γ-glutamyl–peptides inhabituels qui aident à absorber l’excès de glutamate, une autre façon d’atténuer la ferroptose. Tant dans des cellules en culture que chez la souris, la perte de TET1 ou l’inhibition pharmacologique de la synthèse du glutathion a fortement réduit les niveaux de glutathion et de ces peptides protecteurs, rendant les cellules leucémiques beaucoup plus vulnérables aux déclencheurs de la ferroptose.
Une seconde voie d’évasion indépendante de GPX4
De manière surprenante, le rôle protecteur de TET1 ne se limitait pas à l’axe glutathion–GPX4. Même lorsque GPX4 lui-même était supprimée des cellules leucémiques, un excès de TET1 pouvait encore prévenir la mort ferroptotique, suggérant une seconde ligne de défense. Les auteurs ont retracé cela à l’activation par TET1 de la voie de signalisation NFκB, en particulier un composant appelé NFKB2. Cela augmente, à son tour, l’expression de GCH1, une enzyme qui produit la molécule antioxydante BH4. Le BH4 peut protéger les lipides membranaires de l’oxydation sans dépendre de GPX4. Lorsque GCH1 était silencé génétiquement ou bloqué chimiquement, la capacité de TET1 à protéger les cellules de la ferroptose était partiellement perdue. Ensemble, ces résultats définissent une voie TET1–NFKB2–GCH1 qui constitue un système de surveillance de la ferroptose indépendant de GPX4.
Transformer une faiblesse en opportunité thérapeutique
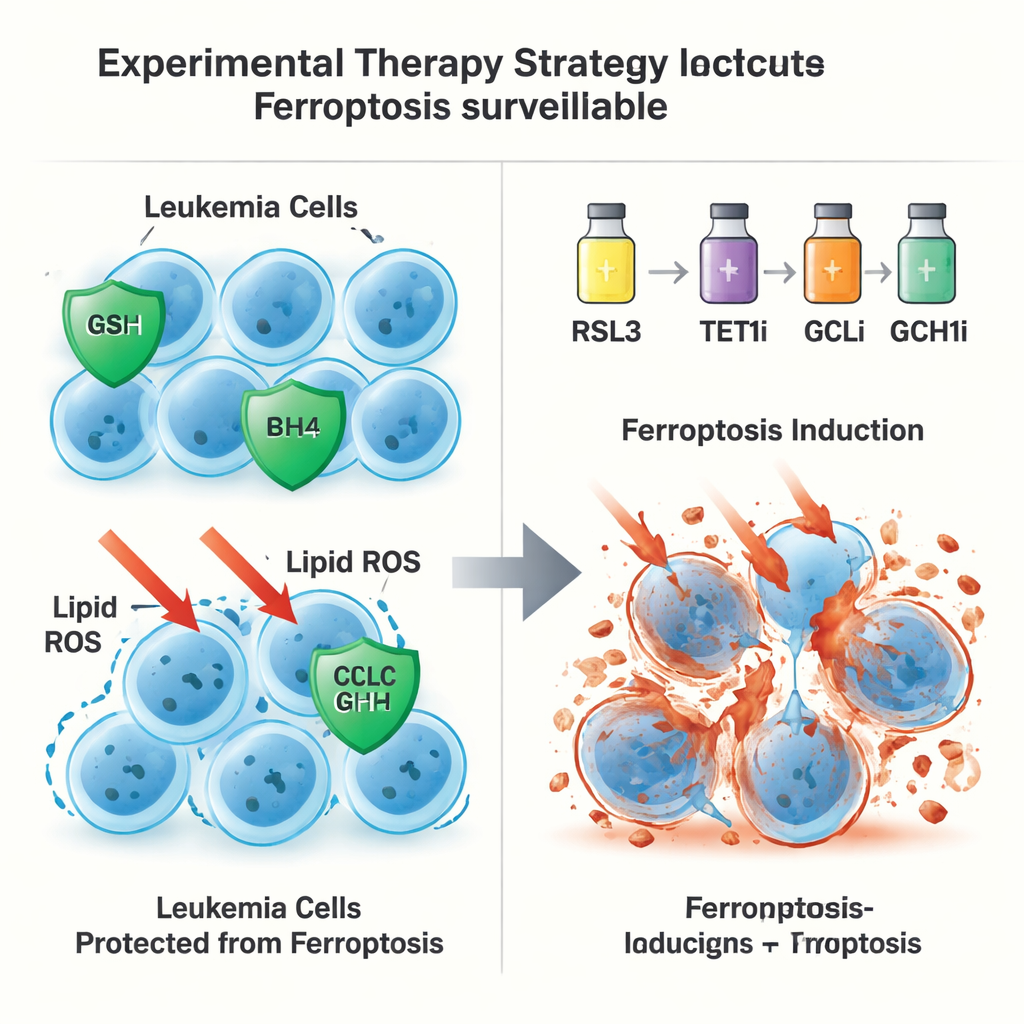
Forts de cette carte à double voie, les chercheurs ont testé si activer la ferroptose tout en affaiblissant les défenses contrôlées par TET1 pouvait offrir un avantage thérapeutique. Dans des modèles murins de LMA et des greffons leucémiques dérivés de patients chez la souris, de faibles doses d’un induceur de ferroptose combinées à des inhibiteurs de TET1, de la synthèse du GSH (via GCLC) ou de GCH1 ont considérablement réduit la charge leucémique, prolongé la survie et épuisé les populations de cellules à l’origine de la leucémie. Fait important, l’inducteur de ferroptose a été utilisé à une fraction des doses rapportées dans des études antérieures, réduisant les préoccupations concernant la toxicité pour les cellules souches sanguines normales.
Ce que cela signifie pour les thérapies anticancéreuses futures
Pour les non-spécialistes, le message clé est que les cellules leucémiques survivent en faisant fonctionner deux systèmes de « bouclier » antioxydant qui se chevauchent, tous deux coordonnés par TET1 : l’un centré sur le glutathion et GPX4, l’autre basé sur GCH1 et BH4. Ce travail montre qu’en activant modestement la ferroptose tout en bloquant TET1 et ses partenaires en aval, les médecins pourraient un jour surmonter la résistance et pousser sélectivement les cellules cancéreuses au-delà du point de non-retour, sans submerger les tissus sains. Bien que ces stratégies ne soient pas encore prêtes pour la clinique, l’étude identifie TET1 comme un nœud de contrôle puissant et une cible prometteuse pour des thérapies combinatoires dans la LMA et potentiellement d’autres cancers difficiles à traiter.
Citation: Yang, L., Lu, J., Yun, W. et al. TET1 as a master regulator controlling GPX4-dependent and -independent ferroptosis surveillance in acute myeloid leukemia. Nat Commun 17, 1800 (2026). https://doi.org/10.1038/s41467-026-68509-x
Mots-clés: ferroptose, leucémie myéloïde aiguë, TET1, glutathion, épigénétique du cancer