Clear Sky Science · fr
Des variantes fonctionnelles en 1p36.23 augmentent le risque de schizophrénie en modulant RERE
Pourquoi de minuscules changements d’ADN comptent pour la santé mentale
La schizophrénie est une maladie mentale grave qui affecte la pensée, les émotions et les relations aux autres. Elle se transmet fortement dans les familles, mais la plupart des modifications génétiques impliquées sont de petites altérations disséminées dans notre ADN. Cette étude se concentre sur l’une de ces régions du génome et montre, pas à pas, comment deux variantes d’ADN subtiles peuvent modifier le développement et la communication des cellules cérébrales de manière à augmenter potentiellement le risque de schizophrénie. 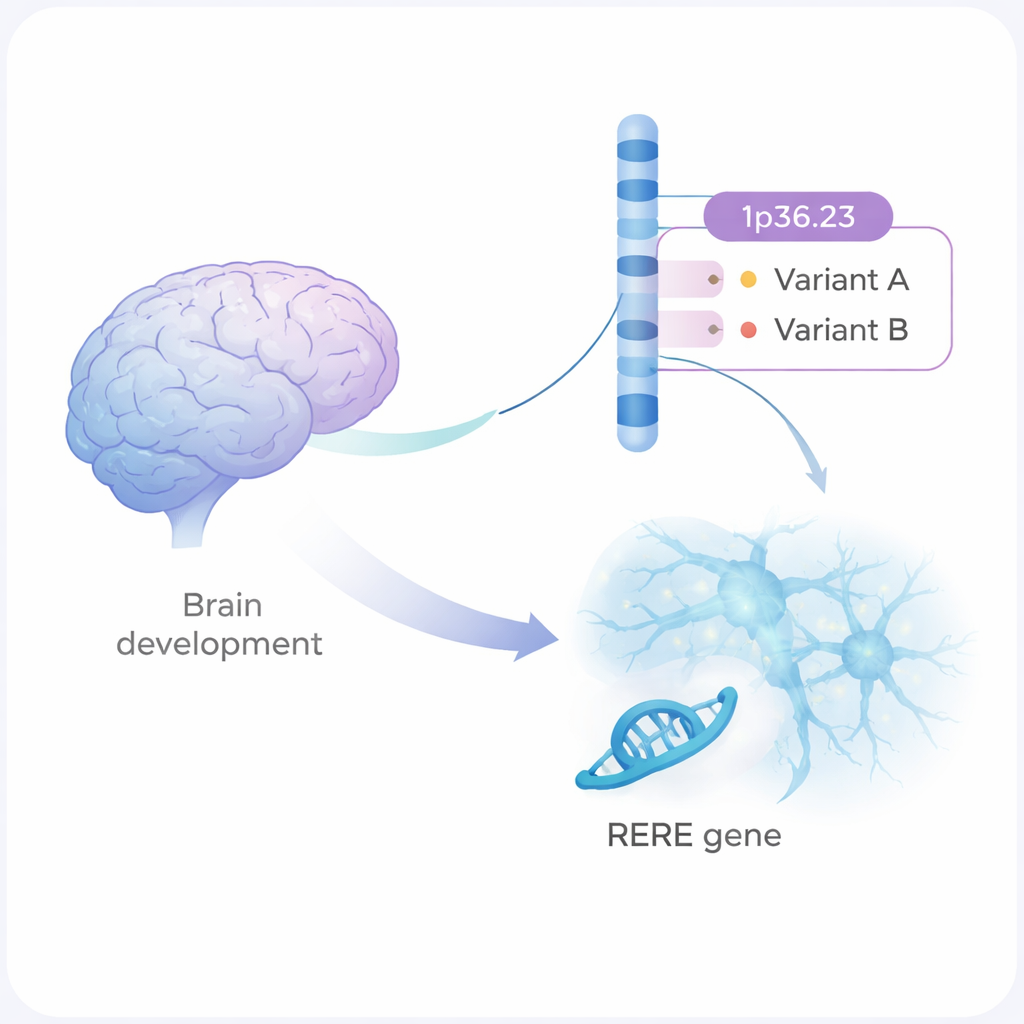
Un point chaud génétique sur le chromosome 1
De larges études génétiques ont mis en évidence plus de 300 régions du génome humain liées à la schizophrénie. L’une d’elles est un segment d’ADN sur le chromosome 1, appelé 1p36.23. Jusqu’à présent, les scientifiques ignoraient quelles modifications exactes de cette région, ni quel gène, étaient réellement en cause. Les auteurs ont combiné la génétique statistique et des expériences de laboratoire et ont identifié deux variantes d’ADN, nommées rs159961 et rs301792, situées à l’intérieur d’un gène appelé RERE. Ces variantes ne modifient pas la protéine RERE elle-même ; elles se trouvent plutôt dans des zones régulatrices — des « interrupteurs » — au sein du gène qui contrôlent l’intensité d’expression de RERE.
Comment les variantes de risque augmentent l’activité de RERE
L’équipe a d’abord cherché à savoir si ces deux variantes agissaient effectivement comme des interrupteurs fonctionnels. À l’aide d’essais rapporteurs — tests où un fragment d’ADN contrôle un gène producteurs de lumière — ils ont montré que les versions associées à la schizophrénie de rs159961 et rs301792 se comportent comme des amplificateurs plus puissants (enhancers) dans des cellules neuronales, mais pas dans des types cellulaires non apparentés. Des tests biochimiques de liaison ont expliqué pourquoi : la forme à risque d’une variante affaiblit l’attachement de REST, une protéine qui freine normalement l’activité génique, tandis que la forme à risque de l’autre renforce la liaison de POLR2A, un élément central de la machinerie de lecture des gènes. Ensemble, ces modifications de la liaison protéine-ADN augmentent l’activité des segments enhancers et élèvent l’expression de RERE.
D’un RERE suractivé à un développement altéré des cellules cérébrales
Puis, les chercheurs ont examiné ce que fait un excès de RERE dans le cerveau. Ils ont constaté que des personnes décédées atteintes de schizophrénie présentaient des niveaux de RERE plus élevés dans leurs tissus cérébraux comparé à des individus non affectés. Pour modéliser cela, ils ont artificiellement augmenté l’expression de RERE dans des cellules souches neurales de souris — les cellules immatures qui donnent naissance aux neurones et aux cellules gliales. Lorsque RERE était surexprimé, ces cellules souches se divisaient moins, restaient bloquées à une phase tardive du cycle cellulaire et produisaient moins de neurones matures, tandis que d’autres types cellulaires restaient en grande partie inchangés. Dans des neurones en culture, un excès de RERE a aussi remodelé leurs ramifications et réduit le nombre et le type de petites protubérances appelées épines dendritiques, où se forment les synapses. Ces modifications s’accordent avec des preuves de longue date montrant que la schizophrénie implique un développement cérébral perturbé et une perte d’épines. 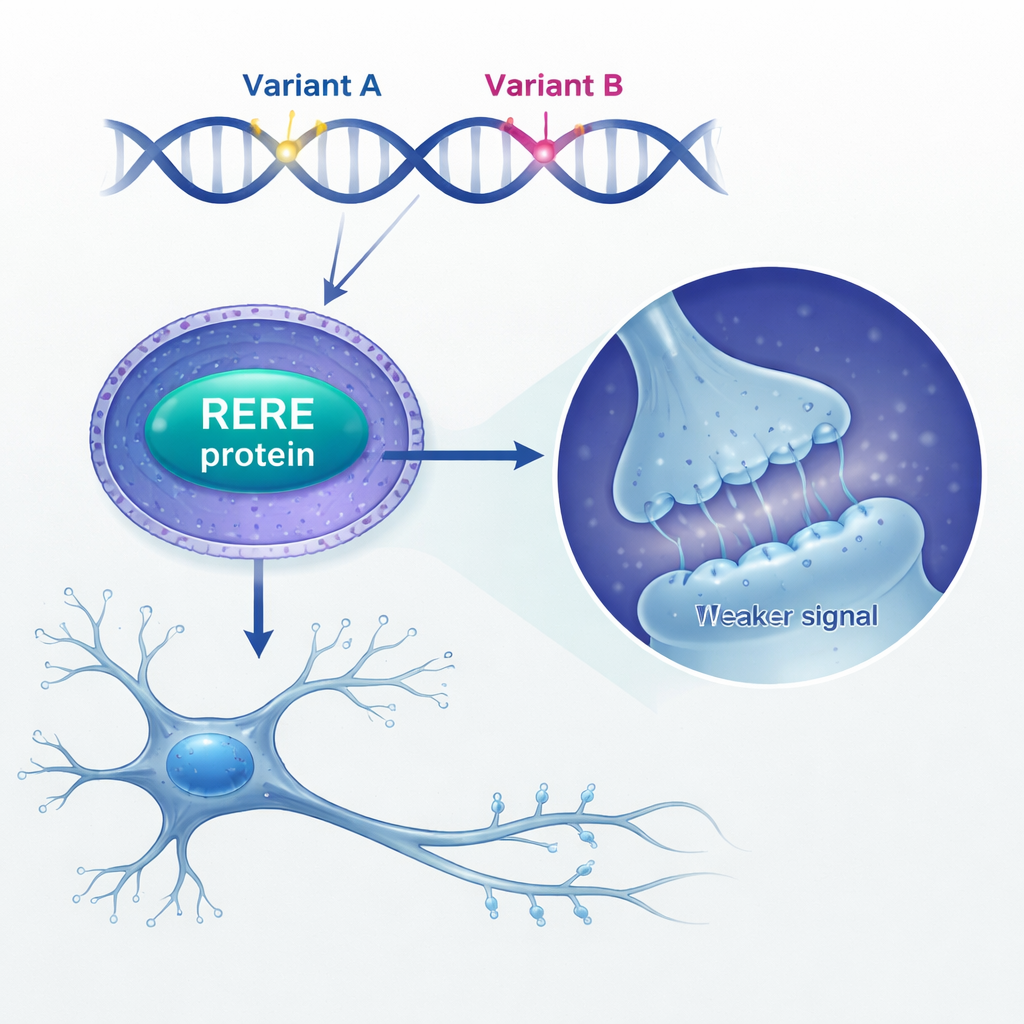
Perturbation de la « conversation » glutamatergique du cerveau
En examinant plus finement l’activité génique, l’équipe a trouvé que la surexpression de RERE perturbait des réseaux de gènes impliqués dans la croissance des dendrites et dans d’importants systèmes de signalisation chimique, en particulier la voie glutamatergique. Une cible clé a émergé : le gène GRIN2A, qui code une sous-unité cruciale (GluN2A) des récepteurs NMDA au glutamate, impliqués depuis longtemps dans la schizophrénie. Les auteurs ont montré que RERE s’associe à deux autres protéines nucléaires, RARB et RXRA, pour se lier directement au promoteur de GRIN2A et en diminuer l’activité. Dans des neurones avec un excès de RERE, les niveaux de GluN2A ont diminué et des enregistrements électrophysiologiques ont révélé des courants synaptiques médiés par les récepteurs NMDA plus faibles, même si la fréquence des événements synaptiques restait inchangée. Autrement dit, le « volume » des signaux excitatoires individuels était réduit.
Relier les variantes d’ADN à la fonction cérébrale
En tissant ensemble génétique, biologie moléculaire, cultures cellulaires et électrophysiologie, ce travail décrit une chaîne causale claire : les variantes d’ADN à risque en 1p36.23 renforcent des éléments régulateurs au sein du gène RERE, conduisant à une expression accrue de RERE dans les cellules cérébrales. L’élévation de RERE altère ensuite la croissance et la maturation des neurones, modifie la forme et le nombre de leurs épines synaptiques, et affaiblit la signalisation glutamatergique via les récepteurs NMDA — en particulier ceux contenant GluN2A. Pour un lecteur non spécialiste, la conclusion est que de très petits changements d’ADN peuvent légèrement modifier l’activité d’un seul gène et que, à l’échelle de nombreuses cellules cérébrales et de nombreuses années, cela peut dévier le développement et la communication cérébrale de manière à contribuer à la schizophrénie.
Citation: Liu, Y., Wang, J., Yang, H. et al. Functional variants at 1p36.23 confer risk of schizophrenia through modulating RERE. Nat Commun 17, 1742 (2026). https://doi.org/10.1038/s41467-026-68449-6
Mots-clés: génétique de la schizophrénie, gène RERE, neurodéveloppement, signalisation glutamatergique, fonction synaptique