Clear Sky Science · fr
Utiliser les références linéaires du pangenome pour découvrir des variants manquants liés à l’autisme
Pourquoi les changements d’ADN invisibles comptent pour l’autisme
La plupart des familles qui sollicitent un test génétique pour un enfant autiste espèrent des réponses claires, mais environ quatre sur cinq n’obtiennent pas d’explication génétique définitive. Cette étude s’attaque à une cause majeure : de nombreux changements d’ADN d’impact sont trop complexes pour les tests standards. En reconstruisant des génomes quasi complets pour 189 personnes issues de 51 familles touchées par l’autisme et en les comparant à une nouvelle référence « pangenome » plus riche, les chercheurs montrent comment des méthodes de séquençage avancées peuvent mettre au jour des mutations rares, jusqu’alors invisibles, susceptibles d’expliquer certains cas d’autisme et d’affections apparentées.
Au-delà des tests génétiques standard
Les tests cliniques traditionnels s’appuient sur de courts fragments d’ADN pour parcourir le génome d’une personne. Cela fonctionne bien pour beaucoup de substitutions d’une seule lettre, mais échoue souvent dans les régions répétitives ou structurellement complexes, précisément là où se cachent certains variants pathogènes puissants. L’équipe s’est concentrée sur des familles pour lesquelles des analyses antérieures en short-read (génome, exome ou panels de gènes) n’avaient pas trouvé la cause de l’autisme ou de symptômes de type Rett. En utilisant le séquençage long-read, qui lit des segments d’ADN beaucoup plus longs, ils ont construit des assemblages de génome phasés et de haute qualité pour 189 individus. Autrement dit, ils ont pu reconstituer les deux copies de chaque chromosome de chaque personne, l’une héritée de chaque parent, avec très peu de lacunes.
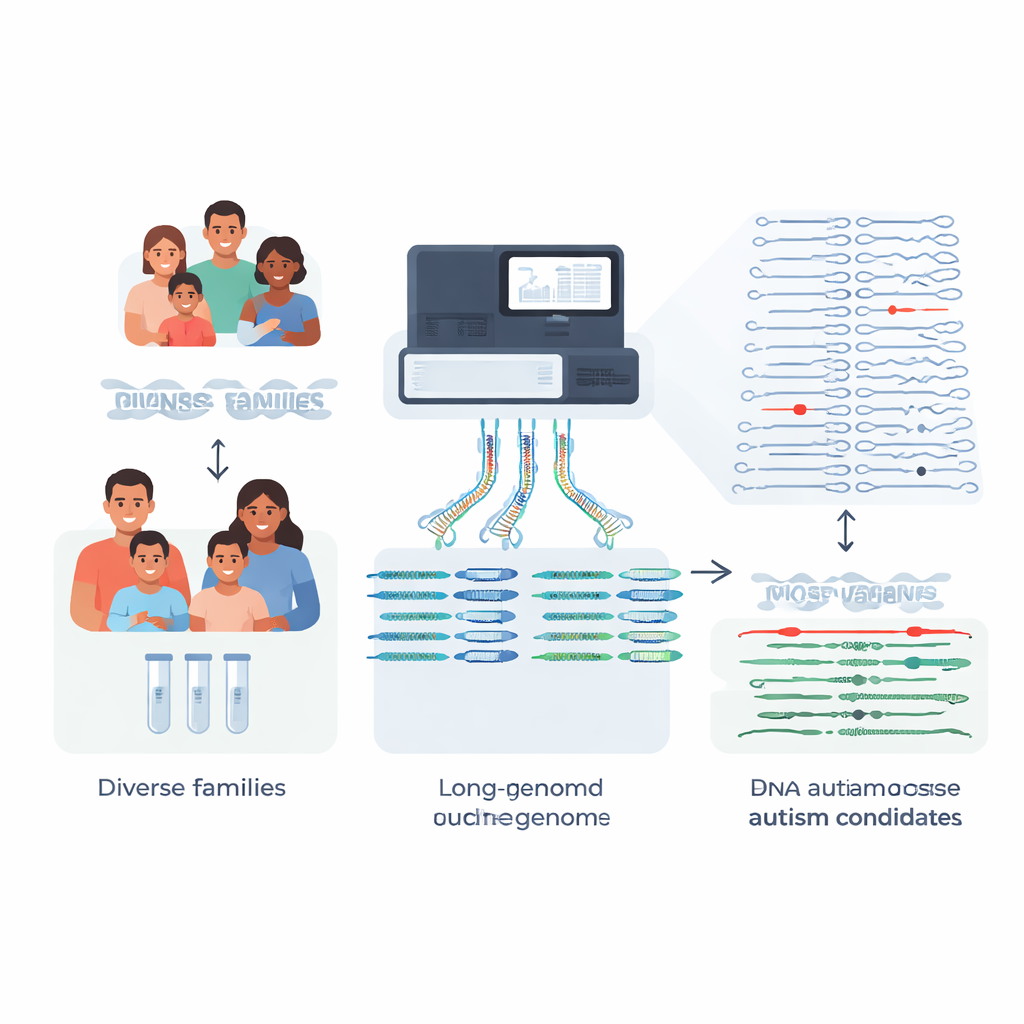
Variants structuraux : des changements majeurs aux effets majeurs
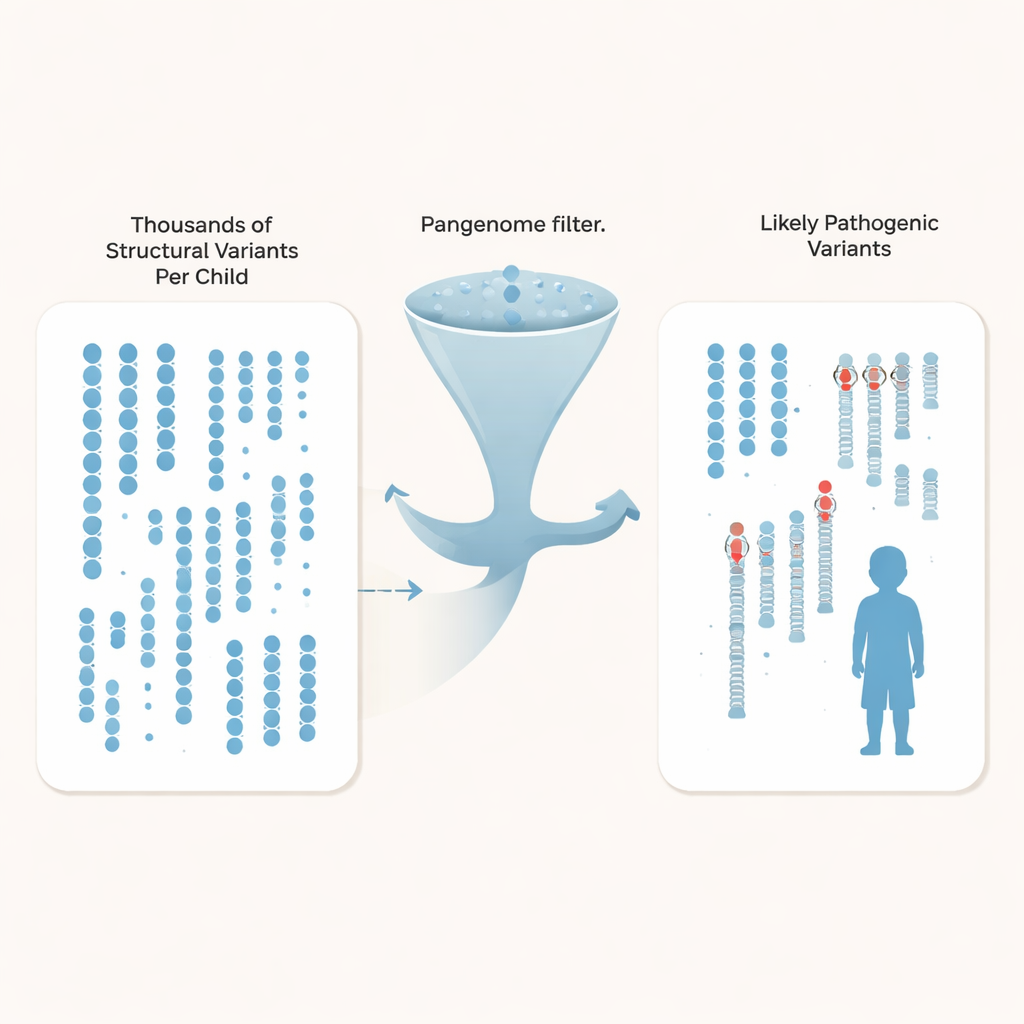
Plutôt que de se limiter aux différences d’une seule lettre, les chercheurs se sont concentrés sur les variants structuraux — insertions, délétions et réarrangements affectant au moins 50 bases d’ADN et pouvant perturber des gènes ou leurs éléments régulateurs. Chaque enfant portait environ 27 000 de ces variants, mais la grande majorité sont des différences bénignes partagées au sein de la population. En comparant leurs familles autistes à des centaines de génomes témoins profondémment séquencés, issus de diverses origines et intégrés au pangenome, l’équipe a pu éliminer plus de 97 % des variants structuraux communs pour chaque enfant, ne laissant qu’environ 600 candidats rares par génome, et parfois aussi peu qu’environ 200 avec l’ensemble de contrôle le plus large.
Retrouver des mutations manquées dans des gènes de risque connus
Avec un espace de recherche drastiquement réduit, les auteurs ont croisé plusieurs lignes de preuve : gènes connus pour l’autisme et les troubles du neurodéveloppement, régions régulatrices actives dans le cortex humain en développement, et schémas de transmission au sein de chaque famille. Ils ont mis au jour trois mutations clairement pathogènes que les tests antérieurs avaient manquées. Cela inclut un nouveau signal d’arrêt dans le gène SYNGAP1, important pour la fonction synaptique, et une délétion qui supprime le dernier exon de MECP2, un gène clé du syndrome de Rett, bien que le patient ait subi plusieurs tests cliniques antérieurs. Ils ont également confirmé une variation causant la maladie dans TBL1XR1, un gène qui interagit avec MECP2. Au total, ils ont signalé neuf autres variants structuraux — souvent transmis et situés dans des régions régulatrices proches de gènes liés au cerveau — comme de solides candidats pour des tests fonctionnels futurs.
Ce que l’étude n’a pas trouvé — et pourquoi cela reste important
Malgré cette recherche approfondie, les auteurs n’ont pas observé d’excès net clair de variants structuraux chez les enfants autistes comparés à leurs frères et sœurs non affectés, du moins dans cette taille d’échantillon modeste. Il y avait toutefois un indice d’un nombre plus élevé de changements structuraux sur le chromosome X chez les filles affectées, et les assemblages quasi complets des chromosomes X et Y leur ont permis de repérer des schémas inhabituels tels qu’un fort biais d’inactivation du X. Ces caractéristiques pourraient devenir des indices importants à mesure que davantage de familles seront étudiées. Surtout, ce travail montre que le séquençage long-read peut récupérer des variants pathogènes que les méthodes short-read manquent, en particulier dans les zones difficiles du génome et dans les régions de contrôle qui modulant l’activité des gènes.
Ce que cela signifie pour les familles et les tests futurs
Pour les familles, l’impact immédiat est modeste mais significatif : parmi ces cas difficiles à élucider, environ 6 % ont reçu un diagnostic génétique clair, et près d’une famille sur cinq a obtenu de nouveaux variants candidats solides à investiguer. Pour le domaine, le message est plus large. À mesure que des références de génomes plus diversifiées et complètes seront ajoutées au pangenome et que le séquençage long-read deviendra plus accessible, les cliniciens pourront exclure rapidement les variants structuraux communs et se concentrer sur un petit ensemble de variants rares et potentiellement nocifs chez chaque patient. Ce changement pourrait peu à peu transformer de nombreux cas d’autisme « non résolus » d’aujourd’hui en cas où la biologie sous-jacente — et les voies possibles de soutien et de traitement — seront bien mieux comprises.
Citation: Sui, Y., Lin, J., Noyes, M.D. et al. Using the linear references from the pangenome to discover missing autism variants. Nat Commun 17, 1681 (2026). https://doi.org/10.1038/s41467-026-68378-4
Mots-clés: génétique de l’autisme, séquençage long-read, variants structuraux, pangenome humain, syndrome de Rett