Clear Sky Science · fr
Conception computationnelle de cyclopropanases généralistes avec sélectivité stéréodivergente
Pourquoi de petites structures à trois anneaux comptent pour les médicaments
Les cyclopropanes — anneaux carbonés à trois atomes — sont de minuscules éléments contrains qui peuvent modifier radicalement le comportement d’un médicament dans l’organisme. L’arrangement tridimensionnel précis des atomes (sa stéréochimie) peut déterminer si une molécule devient un médicament utile ou un analogue inactif, voire nocif. Cet article décrit une stratégie computationnelle pour concevoir des enzymes capables de fabriquer de façon fiable les quatre formes 3D possibles de ces anneaux à partir des mêmes réactifs, ouvrant la voie à une exploration plus rapide et plus propre des candidats-médicaments.
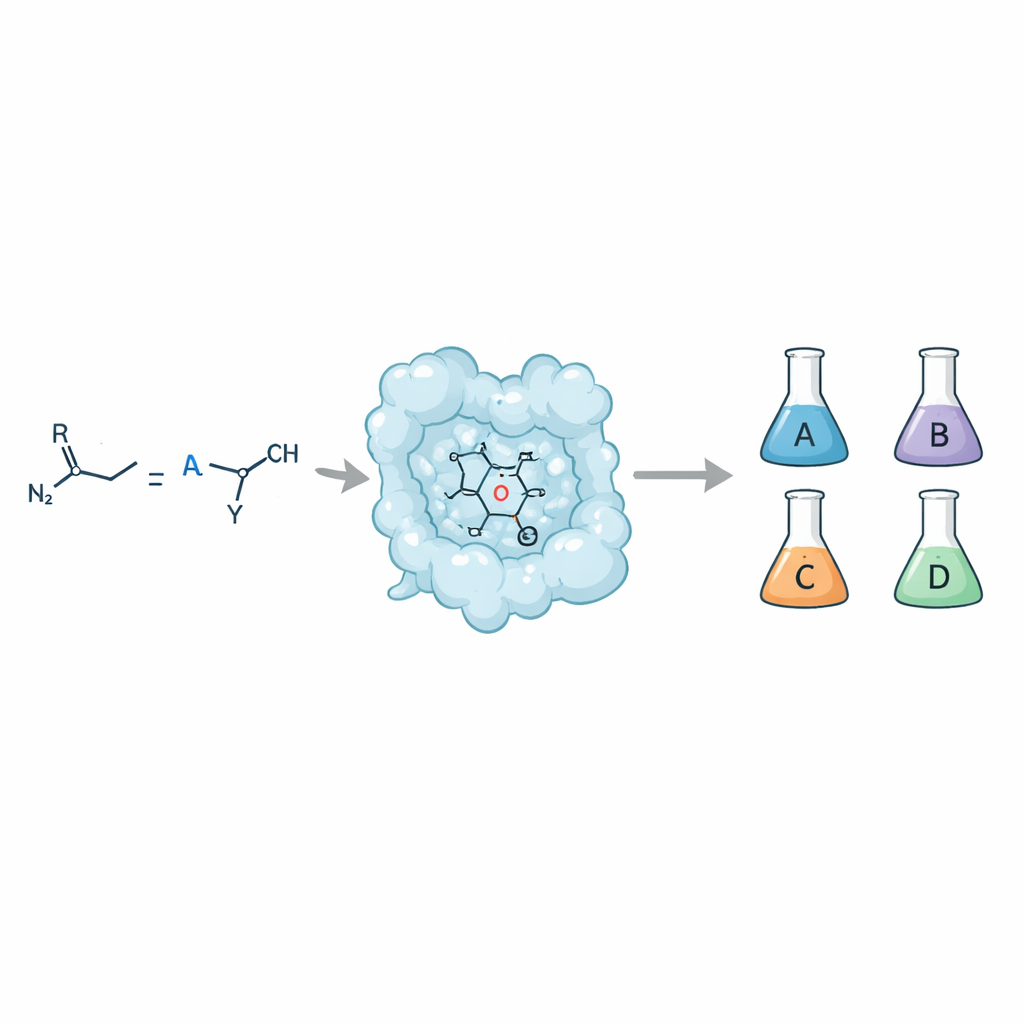
D’une seule recette à quatre résultats différents
Quand une double liaison simple (un oléfine) réagit avec un donneur de carbène tel qu’un composé diazo, le produit peut être un anneau cyclopropane. Mais cet anneau peut adopter quatre formes stéréoisomériques distinctes, toutes constituées des mêmes atomes mais arrangés différemment dans l’espace. Les chimistes veulent accéder à chacune de ces formes car elles interagissent très différemment avec des cibles biologiques et influencent des propriétés clés des médicaments telles que l’absorption, le métabolisme et la sécurité. Les catalyseurs à petite molécule traditionnels peuvent parfois offrir ce contrôle, mais y parvenir avec des enzymes — les catalyseurs naturels — a été difficile, surtout lorsqu’on vise à la fois une forte sélectivité et une large tolérance aux substrats.
Concevoir des enzymes sur écran d’ordinateur
Les auteurs ont développé un flux de travail computationnel multi-états basé sur le mécanisme réactionnel pour résoudre ce problème. Ils ont d’abord utilisé des calculs de chimie quantique pour modéliser les états de transition éphémères — les structures de haute énergie le long du chemin réactionnel — pour la formation de chacune des quatre stéréoisomères de cyclopropane. Ces modèles ont ensuite été placés dans les sites actifs de différentes protéines contenant un hème, et le logiciel de conception protéique Rosetta a été utilisé pour évaluer dans quelle mesure chaque protéine stabilisait ou déstabilisait chaque état de transition. De façon cruciale, le score de conception récompensait les mutations qui favorisaient l’état de transition désiré (conception positive) et défavorisaient les états concurrents (conception négative), apprenant ainsi à l’enzyme à « préférer » un produit 3D plutôt qu’un autre.
Construire une boîte à outils enzymatique complète
Avec cette approche, l’équipe a créé une famille d’enzymes « généralistes » cyclopropanases. À partir de la myoglobine, ils ont repensé le site actif pour obtenir des variants qui produisent le cyclopropane trans-(1R,2R) avec une très haute sélectivité et une bonne activité sur plus de 20 oléfines différentes, y compris des substrats peu réactifs ou pauvres en électrons. Une myoglobine préalablement conçue fournissait déjà le produit complémentaire trans-(1S,2S). Pour obtenir les deux produits cis, les auteurs se sont tournés vers d’autres protéines hémées. Ils ont remodelé l’enzyme bactérienne P450cam pour obtenir des variants donnant sélectivement le produit cis-(1S,2R), et ils ont détourné l’enzyme humaine indoleamine 2,3‑dioxygénase‑1 (IDO1) — jusque-là non utilisée pour la chimie des carbènes — pour favoriser le produit cis-(1R,2S). Au total, ces quatre biocatalyseurs peuvent fournir chaque stéréoisomère du même jeu de produits cyclopropanes, souvent avec un contrôle atteignant 99 % à la fois sur la diastéréomérie et l’énantiosélectivité.
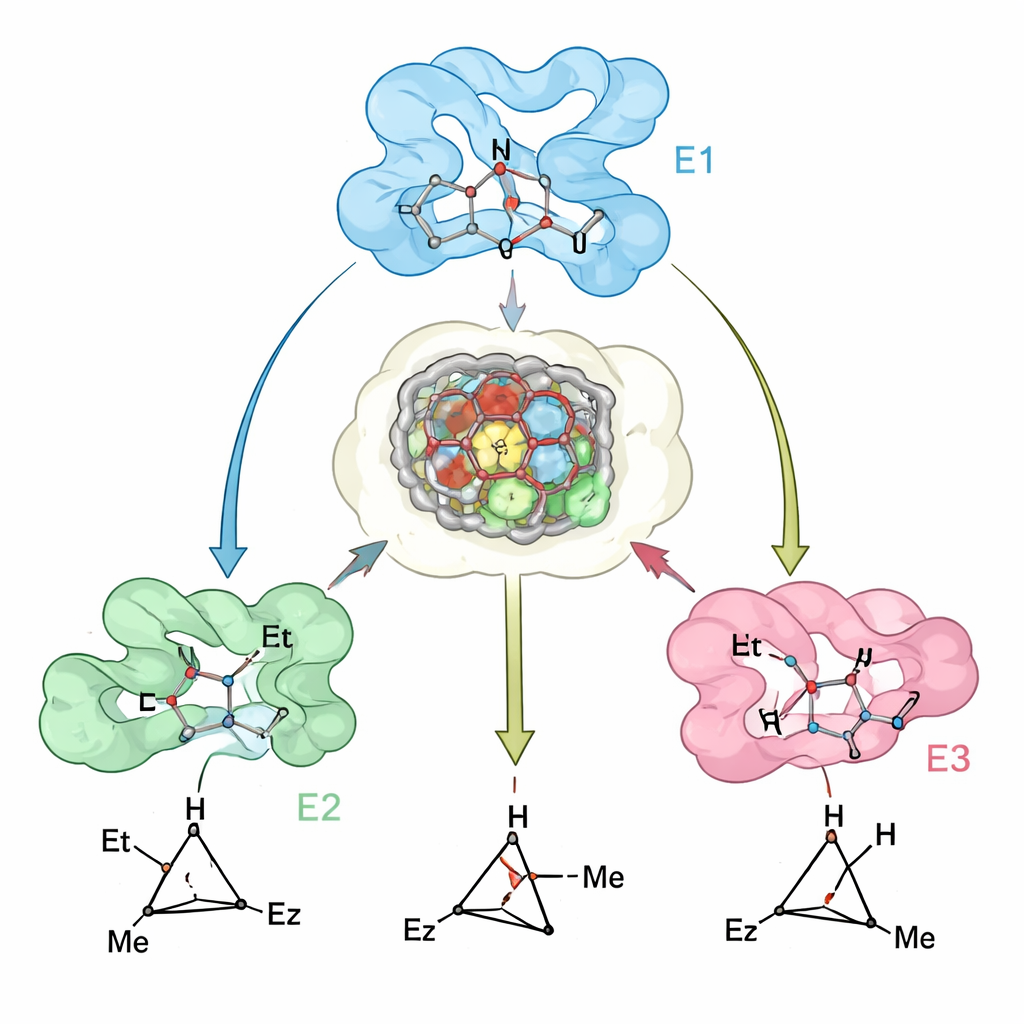
Voir comment la conception correspond à la réalité
Pour vérifier à quel point leurs modèles computationnels reflétaient les enzymes réelles, les chercheurs ont résolu des structures cristallines d’un variant clé de myoglobine et les ont comparées aux structures prédites. L’accord était étroit, et les données expérimentales ont mis en évidence une caractéristique subtile mais importante : le site actif de la protéine est pré-organisé pour accueillir l’état de transition préféré, tandis que de petits déplacements dans des boucles et des hélices voisines rendent la liaison de l’état de transition « erroné » énergétiquement défavorable. Lorsque les prédictions étaient moins exactes — par exemple pour quelques substrats volumineux — les divergences pouvaient être attribuées à des mouvements de la charpente (backbone) qui n’avaient pas été entièrement capturés dans la modélisation, indiquant des pistes claires pour améliorer les méthodes de conception futures.
Ce que cela implique pour les futurs médicaments et catalyseurs
En combinant la modélisation des états de transition fondée sur la physique avec une refonte protéique intelligente, ce travail démontre que les issues stéréochimiques des réactions catalysées par des enzymes peuvent être programmées à l’avance, plutôt que découvertes uniquement par évolution itérative et tâtonnements. La suite de cyclopropanases obtenue offre aux chimistes un moyen pratique de produire des jeux complets de stéréoisomères de cyclopropane à partir d’un large éventail d’oléfines de départ, simplifiant grandement les études structure‑activité en découverte de médicaments et en synthèse de produits naturels. La même stratégie devrait être adaptable à d’autres types d’enzymes et classes de réactions, accélérant la création de biocatalyseurs offrant un contrôle 3D précis sur des molécules complexes.
Citation: Shen, Z., Siriboe, M.G., Ren, X. et al. Computational design of generalist cyclopropanases with stereodivergent selectivity. Nat Commun 17, 1620 (2026). https://doi.org/10.1038/s41467-026-68327-1
Mots-clés: biocatalyse, cyclopropanation, conception d'enzymes, stéréochimie, protéines hémées