Clear Sky Science · fr
Des variants de novo dans le gène du facteur d’épissage SF3B1 sont associés à des troubles du développement neurologique
Quand un seul gène perturbe le plan précoce du cerveau
Pourquoi certains enfants développent-ils des difficultés d’apprentissage, des crises d’épilepsie ou des troubles de l’alimentation alors que la grossesse et l’accouchement semblaient normaux ? Cette étude examine un gène unique, nommé SF3B1, qui aide les cellules à traiter les messages génétiques. Les chercheurs montrent que des changements nouveaux et spontanés de ce gène peuvent perturber subtilement la manière dont les cellules cérébrales lisent les instructions de l’ADN, conduisant à un syndrome neurodéveloppemental jusqu’ici méconnu.
Un maître-éditeur des messages génétiques
Chaque cellule de notre corps doit convertir le texte génétique brut en instructions claires avant de pouvoir fabriquer des protéines. Cette étape d’édition, appelée épissage de l’ARN, élimine les segments non codants et recoud les parties utiles. SF3B1 est un élément central de la « machine » d’épissage cellulaire. Jusqu’à présent, les altérations de SF3B1 étaient surtout connues pour leur rôle dans les cancers, où les cellules tumorales acquièrent des mutations de ce gène au cours de la vie. Le travail présent pose une question différente : que se passe-t-il quand une altération délétère de SF3B1 est présente dès la conception dans toutes les cellules de l’organisme ?
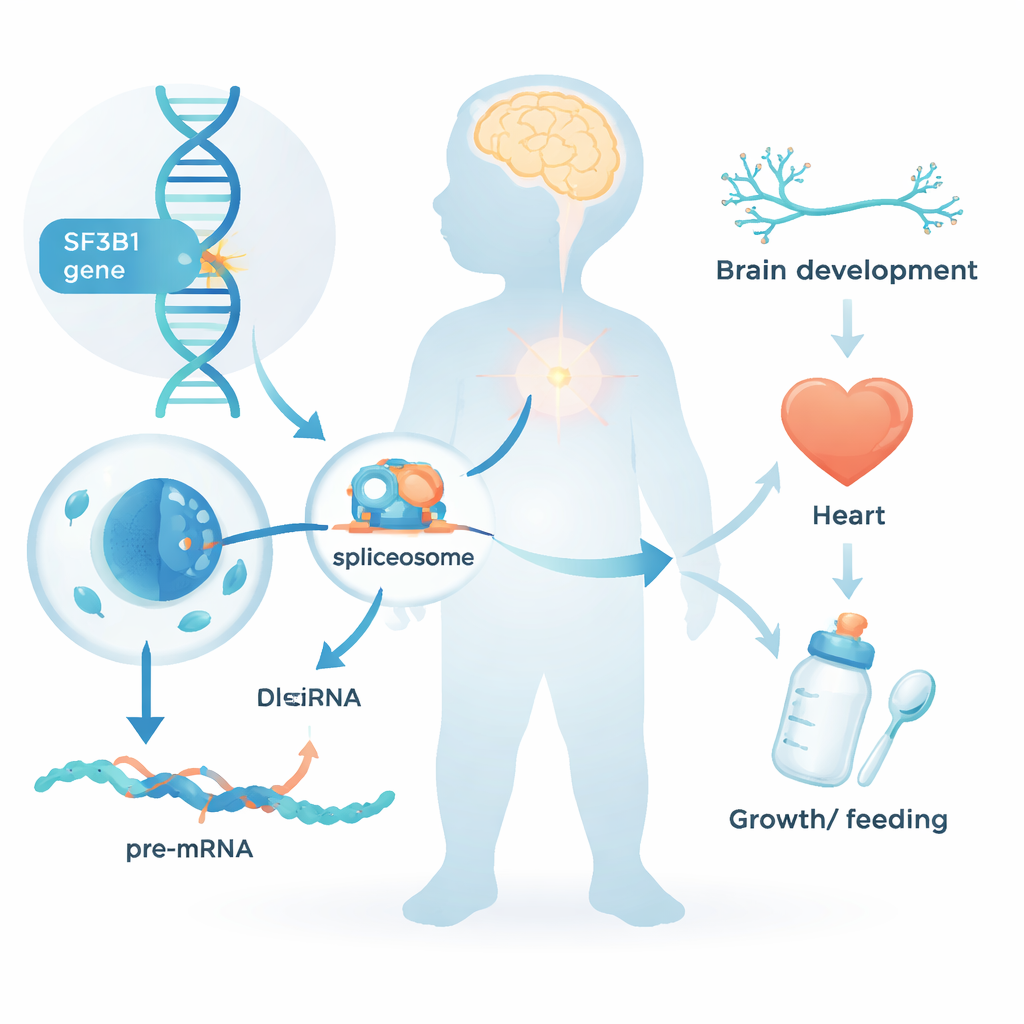
Un syndrome infantile nouvellement reconnu
L’équipe a rassemblé des données provenant de 26 enfants et jeunes adultes porteurs de variants rares de SF3B1, survenant pour la plupart de novo — c’est‑à‑dire non hérités des parents. Presque tous présentaient des troubles du développement neurologique : retards pour s’asseoir, marcher ou parler ; déficience intellectuelle le plus souvent légère à modérée ; et, dans environ la moitié des cas, des crises d’épilepsie. Beaucoup avaient un tonus musculaire faible et nécessitaient une aide pour l’alimentation, parfois via une gastrostomie. Les traits faciaux étaient subtilement atypiques mais variables d’un enfant à l’autre ; un caractère frappant et fréquent était un palais haut ou fendu. Plusieurs participants présentaient aussi des malformations cardiaques, un retard de croissance ou une petite taille crânienne, montrant que l’impact des altérations de SF3B1 dépasse le seul cerveau.
Deux classes de changements génétiques, deux profils cliniques
Les chercheurs ont distingué deux grands types de variants de SF3B1. Un groupe comprenait des altérations « perte de fonction », comme des signaux d’arrêt prématuré, qui devraient réduire la quantité de protéine SF3B1 fonctionnelle. Le second groupe contenait des variants faux-sens, où un seul acide aminé de la protéine est modifié. En regroupant les caractéristiques médicales des enfants, l’équipe a observé que ceux porteurs de variants faux-sens avaient tendance à présenter des troubles plus graves et plus complexes, incluant un taux plus élevé d’anomalies cardiaques et gastro-intestinales, une petite taille et une microcéphalie. Les variants perte de fonction, en revanche, étaient parfois hérités d’un parent légèrement atteint voire apparemment sain, suggérant que la simple diminution de SF3B1 peut être compatible avec des symptômes relativement bénins chez certains individus.
Des erreurs de réglage fin plutôt qu’une défaillance totale
Pour comprendre l’action des variants faux-sens dans les cellules, les scientifiques les ont recréés dans des lignées cellulaires en laboratoire. Contre toute attente, ces protéines SF3B1 altérées pouvaient encore accomplir suffisamment la tâche d’épissage pour compenser des cellules dont le SF3B1 normal avait été éteint. Cela a écarté une explication simple de type perte de fonction. Par séquençage profond de l’ARN, l’équipe a ensuite examiné l’ensemble des messages cellulaires. Ils ont constaté que les variants faux-sens modifiaient subtilement l’épissage de centaines de gènes, notamment en changeant le choix des sites d’épissage aux extrémités des exons et en provoquant parfois l’exclusion d’exons. L’ampleur de la perturbation était moindre que celle observée avec la mutation classique associée au cancer K700E, mais néanmoins significative : de nombreux gènes affectés interviennent dans le développement du cerveau, le câblage neuronal et des processus fondamentaux comme la gestion de l’ARN et la synthèse protéique.
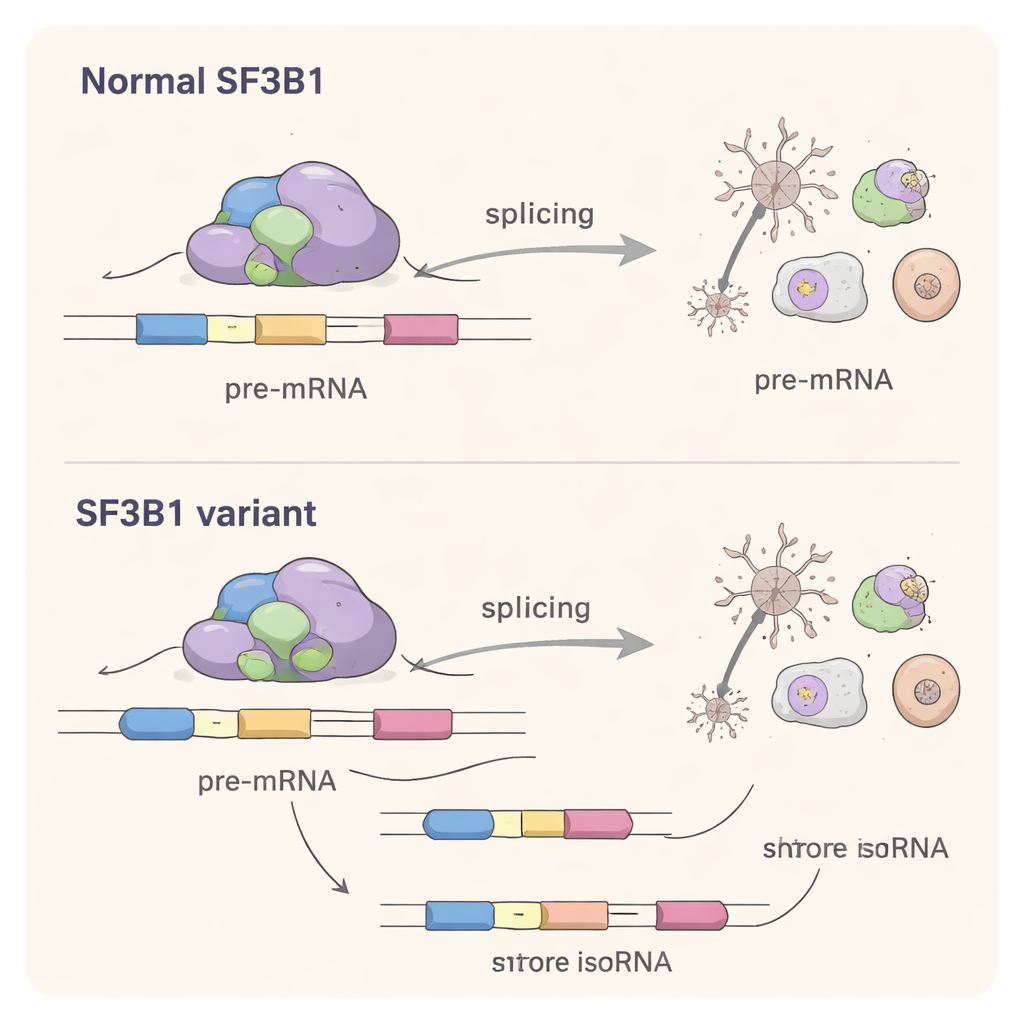
Un mécanisme commun entre cancer et troubles cérébraux
Bien que la plupart des variants neurodéveloppementaux de SF3B1 se situent à des positions différentes des mutations bien connues en cancérologie, ils perturbent le même processus central : la reconnaissance précise des sites d’épissage dans l’ARN. L’étude montre que ces variants du développement possèdent leur propre « signature d’épissage », choisissant des sites alternatifs souvent plus proches des sites normaux que ceux favorisés dans le cancer. Cela suggère un mécanisme de changement de fonction, où la protéine mutée entre en compétition avec la copie normale et pousse la machinerie d’épissage vers des choix légèrement erronés dans de nombreux gènes simultanément.
Ce que cela signifie pour les familles et la recherche future
Pour les familles concernées, ce travail identifie SF3B1 comme une nouvelle cause de troubles du développement neurologique qui peut désormais être détectée en consultation génétique, offrant potentiellement la fin de longues recherches diagnostiques. Plus largement, il ajoute SF3B1 à une liste restreinte mais croissante de gènes d’épissage dont les altérations peuvent provoquer à la fois le cancer et des troubles cérébraux infantiles, selon le moment et la nature de la modification. En cartographiant comment des variants spécifiques de SF3B1 reconfigurent l’épissage de l’ARN, l’étude prépare le terrain pour de futures thérapies visant à corriger de manière ciblée ces erreurs d’épissage.
Citation: Uguen, K., Bergot, T., Scott-Boyer, MP. et al. De novo variants in the splicing factor gene SF3B1 are associated with neurodevelopmental disorders. Nat Commun 17, 1569 (2026). https://doi.org/10.1038/s41467-026-68284-9
Mots-clés: Épissage de l’ARN, SF3B1, troubles du développement neurologique, variants de novo, spliceosomopathies