Clear Sky Science · fr
Bonnes pratiques et outils en R et Python pour le traitement statistique et la visualisation des données en lipidomique et métabolomique
Pourquoi il est important de transformer des chiffres de laboratoire en images claires
Les instruments modernes peuvent désormais mesurer des milliers de petites molécules — lipides et autres métabolites — dans une seule goutte de sang ou de tissu. Ces mesures contiennent des indices sur les risques de maladie, les réponses aux traitements et la façon dont notre organisme réagit à l’alimentation ou au vieillissement. Mais la sortie brute n’est pas une réponse prête à l’emploi : il s’agit d’un énorme tableau de nombres qui doit être nettoyé, analysé et transformé en représentations compréhensibles. Cet article explique comment les chercheurs peuvent utiliser deux langages de programmation populaires, R et Python, pour le faire de manière fiable, transparente et avec des graphiques de qualité publication.
Des mesures chimiques à des tableaux de données complexes
En lipidomique et métabolomique, la spectrométrie de masse et la chromatographie génèrent de grands ensembles de données où chaque ligne représente un échantillon et chaque colonne une molécule. Ces tableaux ne se comportent rarement comme des exemples pédagogiques bien rangés. Ils contiennent des valeurs manquantes, des valeurs aberrantes et des distributions asymétriques où quelques molécules présentent des niveaux extrêmement élevés. Les concentrations peuvent s’étendre sur plusieurs ordres de grandeur et être influencées par l’âge, le sexe, l’alimentation, les médicaments, les rythmes circadiens et des problèmes techniques tels que la dérive d’instrument ou les effets de lot. Des groupes d’experts internationaux ont émis des recommandations pour standardiser la collecte, le traitement et la déclaration des échantillons, mais même avec de bonnes pratiques de laboratoire, un traitement statistique soigneux reste essentiel pour extraire des signaux biologiques véritables de ce fond bruyant.
Nettoyer et préparer les nombres
Avant qu’une comparaison entre groupes sains et malades ait du sens, les données doivent être préparées. La revue décrit comment apparaissent les valeurs manquantes — par des aléas, des limites instrumentales ou des interférences de signal — et explique quand elles peuvent être ignorées sans risque, quand elles doivent être ré‑mesurées, et comment elles peuvent être estimées de façon raisonnable (imputées) à l’aide de méthodes telles que les k-plus proches voisins, les forêts aléatoires ou la substitution par de faibles valeurs. Ensuite, les auteurs présentent des stratégies de normalisation qui réduisent la variation indésirable, par exemple en corrigeant les effets de lot avec des échantillons de contrôle de qualité ou en ajustant les différences de quantité d’échantillon. Ils discutent ensuite des transformations comme le logarithme — qui apprivoise les longues queues à droite des distributions — et des méthodes de mise à l’échelle qui mettent toutes les molécules sur un pied d’égalité afin que les composés très variables ne dominent pas les analyses ultérieures. 
Tests statistiques et récits visuels
Une fois les données correctement préparées, une palette d’outils statistiques entre en jeu. Pour une molécule unique, les chercheurs peuvent calculer des rapports de variation (fold change) et utiliser des tests classiques comme le test t ou leurs équivalents non paramétriques (comme le test de Mann–Whitney) pour déterminer si les niveaux diffèrent entre groupes. Pour des comparaisons impliquant plusieurs groupes, des méthodes telles que l’ANOVA ou le test de Kruskal–Wallis sont présentées, accompagnées de procédures post hoc pour identifier précisément quelles paires de groupes diffèrent. La puissance de ces tests se révèle lorsque leurs résultats sont visualisés clairement. L’article met en avant les boîtes à moustaches (y compris des variantes améliorées pour les données asymétriques), les violons et les graphiques en volcan qui combinent taille d’effet et signification statistique. Pour les lipides, des visuels plus spécialisés sont décrits, comme les réseaux lipidiques montrant des changements coordonnés au sein de classes complètes, et les graphiques des chaînes d’acyle gras révélant des motifs de longueur de chaîne carbonée et de saturation.
Voir des motifs dans de nombreuses variables à la fois
Puisque chaque échantillon peut comporter des centaines ou des milliers de molécules mesurées, les méthodes multivariées sont cruciales. La revue explique comment l’analyse en composantes principales (ACP) compresse cette complexité en quelques nouveaux axes capturant les principales directions de variation, permettant des vérifications rapides de la séparation entre groupes, des effets de lot ou de la stabilité analytique. Des méthodes non linéaires plus avancées, incluant t‑SNE et UMAP, peuvent révéler des grappes et des structures subtiles dans l’espace de haute dimension. Dans les situations où l’objectif est de classifier des échantillons — par exemple distinguer des patients de témoins — les auteurs décrivent des approches supervisées basées sur les moindres carrés partiels et son extension orthogonale (PLS‑DA et OPLS‑DA). Ces méthodes lient les profils moléculaires aux étiquettes d’échantillon, facilitent la sélection de variables et sont souvent résumées par des graphiques de scores, des graphiques de charges et des courbes ROC.
Boîtes à outils pratiques en R et Python
Pour aider les débutants à passer de la théorie à la pratique, l’article passe en revue un vaste écosystème de paquets logiciels. En R, des collections comme tidyverse et tidymodels simplifient la manipulation des données et la modélisation, tandis que ggplot2 et des paquets complémentaires tels que ggpubr, ggstatsplot et tidyplots facilitent la production de figures prêtes pour la publication. Des bibliothèques spécialisées prennent en charge l’ACP, le clustering et les modèles basés sur PLS, et les paquets Bioconductor offrent des cartes thermiques complexes et des graphiques interactifs. En Python, pandas assure la gestion de tableaux, tandis que matplotlib, seaborn et plotly couvrent la visualisation, et scikit‑learn propose une large gamme de méthodes multivariées. Tout au long de l’article, les auteurs insistent sur des exemples pas à pas disponibles dans un GitBook d’accompagnement, afin que les lecteurs puissent reproduire les workflows et les adapter à leurs propres données. 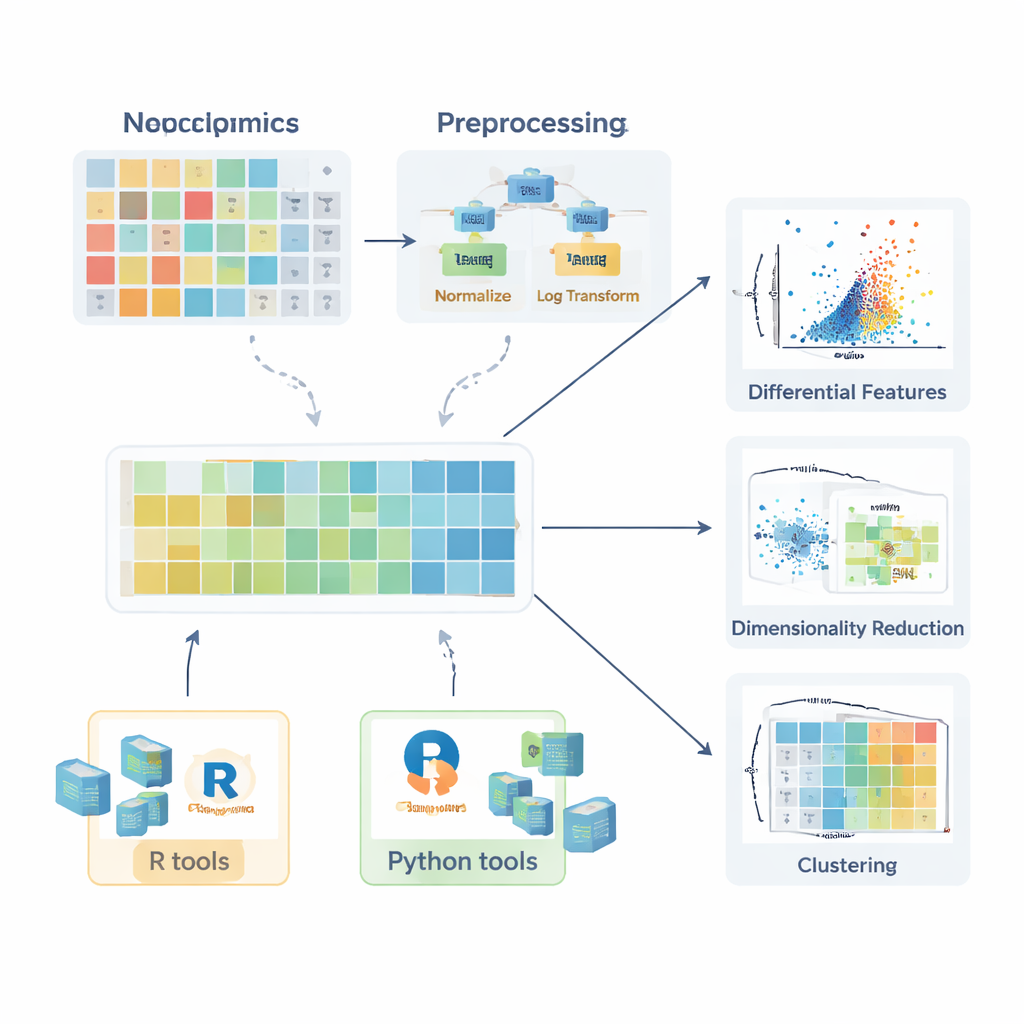
Transformer une chimie complexe en informations fiables
L’article conclut que la véritable promesse de la lipidomique et de la métabolomique réside non seulement dans la puissance des instruments, mais dans la manière dont leurs résultats sont traités et visualisés de manière réfléchie. En suivant de bonnes pratiques statistiques, en utilisant des outils ouverts et bien documentés en R et Python, et en s’appuyant sur des exemples de code partagés, les chercheurs peuvent construire des pipelines robustes et reproductibles. Cela augmente les chances que les motifs identifiés parmi de minuscules molécules se traduisent en biomarqueurs fiables, en une meilleure compréhension des mécanismes de maladie et en approches plus personnalisées de la médecine, au bénéfice ultime des patients.
Citation: Idkowiak, J., Dehairs, J., Schwarzerová, J. et al. Best practices and tools in R and Python for statistical processing and visualization of lipidomics and metabolomics data. Nat Commun 16, 8714 (2025). https://doi.org/10.1038/s41467-025-63751-1
Mots-clés: lipidomique, métabolomique, visualisation de données, programmation R, Python