Clear Sky Science · fr
Combiner la transcriptomique spatiale avec la morphologie tissulaire
Observer les tissus de deux manières complémentaires
Les médecins et les chercheurs veulent de plus en plus savoir non seulement quels gènes sont actifs dans un tissu, mais aussi précisément où ils sont exprimés. Parallèlement, les microscopes des hôpitaux capturent déjà quotidiennement des images riches de la structure tissulaire que les pathologistes utilisent. Cet article explique comment un nouveau domaine cherche à relier ces deux perspectives — des cartes détaillées de l'activité génique et des images de microscope classiques — et pourquoi cette combinaison pourrait permettre des diagnostics plus précoces, un meilleur classement des cancers et une compréhension plus fine de la façon dont les maladies se développent et se propagent.
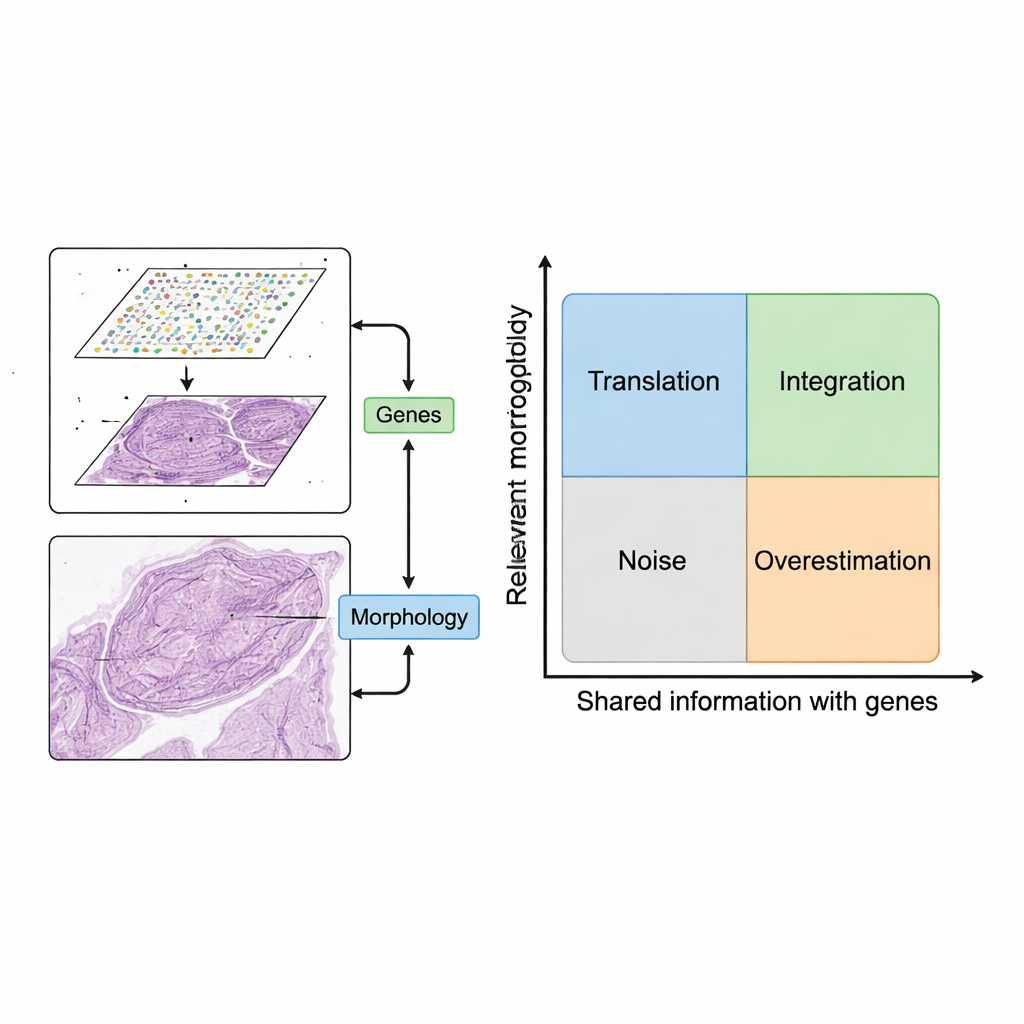
Des cellules dispersées aux cartes d'activité génique
Pendant des années, les puissantes méthodes « omiques » nécessitaient de broyer les tissus en un mélange de cellules individuelles, ce qui détruisait l'information sur l'origine spatiale de chaque cellule. La transcriptomique spatiale a changé la donne en mesurant l'activité génique tout en conservant la position de chaque cellule dans le tissu. Le résultat est une grille de points, chacun associé à un profil d'expression génique et à des coordonnées précises. À elle seule, cette donnée spatiale a déjà révélé de nouveaux schémas de diversité cellulaire et d'architecture des maladies. Mais elle est figée au moment de la mesure, et répéter l'expérience coûte cher. En revanche, les images tissulaires colorées avec des teintures standard, comme l'hématoxyline et l'éosine (H&E) largement utilisées, sont peu coûteuses et abondantes, et contiennent des indices visuels sur la forme des cellules, leur densité et l'organisation tissulaire.
Deux façons de combiner images et gènes
La revue propose un cadre simple mais puissant pour utiliser ces deux sources de données ensemble. D'abord, des patchs d'image sont appariés avec des points d'expression génique à proximité. Ensuite, des modèles informatiques extraient des caractéristiques des images — des motifs capturant la forme, la texture et l'organisation — et les comparent avec les motifs d'expression génique. Les auteurs décrivent deux scénarios souhaitables. Dans la « traduction », les caractéristiques d'image suivent étroitement l'activité génique pertinente, permettant aux modèles de prédire quels gènes sont exprimés à partir de l'image seule. Cela peut servir à combler des mesures géniques manquantes, atteindre une résolution plus fine que la grille d'origine ou inférer l'activité génique à partir de lames cliniques routine sans analyses supplémentaires. Dans l'« intégration », les caractéristiques d'image captent des informations utiles que les données géniques manquent, comme des changements structurels lents ou une organisation tissulaire subtile, aidant à définir des régions ou « domaines » plus clairs au sein d'un tissu.
Quand l'information supplémentaire aide — et quand elle nuit
Toute caractéristique d'image n'est pas pertinente. Les auteurs introduisent une carte conceptuelle à deux axes : dans quelle mesure une caractéristique d'image est-elle pertinente pour la question biologique, et dans quelle mesure elle recoupe l'information génique. Les caractéristiques qui ne sont ni pertinentes ni liées aux gènes ne sont que du bruit, comme des artefacts de coloration. Des caractéristiques qui suivent des motifs géniques mais concernent des gènes peu importants (par exemple des gènes de ménage de base) peuvent donner l'impression de bonnes performances tout en apportant peu de valeur clinique. En organisant les méthodes en quatre quadrants — traduction, intégration, bruit et surestimation — le cadre clarifie quand la combinaison d'images et de gènes apporte réellement un aperçu nouveau, et quand elle ne fait que répéter ou masquer ce qui est déjà connu.
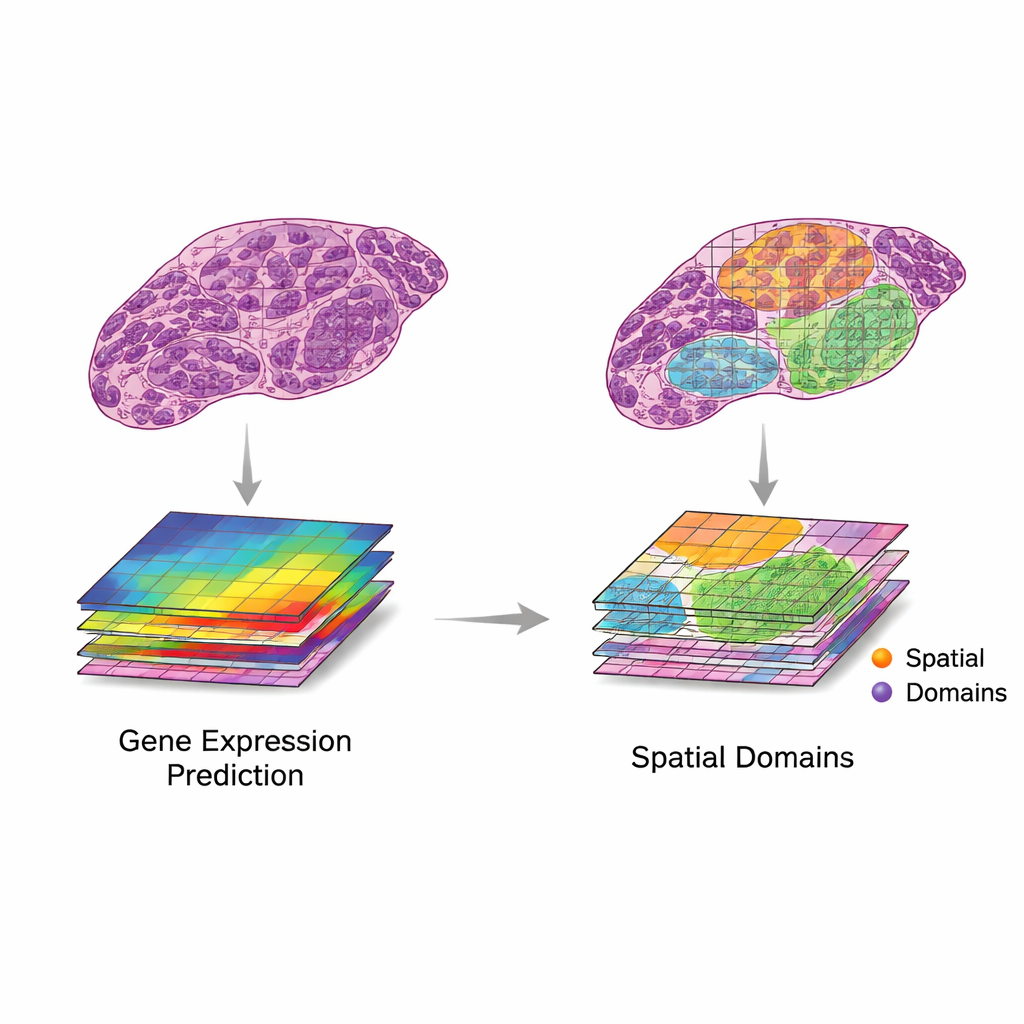
Outils actuels, tests et difficultés croissantes
Une vague rapide de méthodes d'intelligence artificielle tente désormais d'effectuer traduction et intégration sur des données réelles. Les premiers systèmes s'appuyaient sur des réseaux de neurones convolutionnels, tandis que les plus récents utilisent des transformers, des réseaux de neurones graphiques et des modèles multi‑échelles capables de capter des détails allant de minuscules structures cellulaires au contexte de la lame entière. Ces méthodes ont été utilisées pour prédire l'activité génique à partir d'images H&E, générer des cartes à super‑résolution et aider à identifier des régions tissulaires au comportement distinct. Pour juger de la performance, les chercheurs s'appuient sur des mesures statistiques comme la corrélation entre niveaux géniques prédits et observés, ou l'accord entre régions définies par l'IA et annotations d'experts pathologistes. Cependant, les jeux de données restent petits et hétérogènes, et la comparaison entre études est difficile. De nombreux gains rapportés peuvent refléter un surapprentissage, ou des succès sur des gènes et motifs peu pertinents en clinique.
Vers quoi cela peut conduire
Les auteurs concluent que la combinaison de cartes géniques spatiales et d'images tissulaires est prometteuse mais encore à un stade précoce. Les modèles actuels atteignent souvent une précision modérée et ne sont pas encore prêts pour un usage médical courant. Les progrès futurs viendront probablement de meilleures caractéristiques d'image, en particulier de grands « modèles fondamentaux » entraînés sur des millions de lames de pathologie, et d'une focalisation sur les gènes et motifs qui influent réellement sur la prise en charge des patients. Une intégration soigneusement conçue pourrait un jour révéler des signaux d'alerte précoces en repérant des discordances entre l'apparence actuelle d'un tissu et ce que ses gènes prédisent pour l'avenir. En bref, ce travail trace une feuille de route pour transformer des images microscopiques de routine en cartes riches, informées par la génétique, qui aident les médecins à comprendre et traiter les maladies avec plus de précision.
Citation: Chelebian, E., Avenel, C. & Wählby, C. Combining spatial transcriptomics with tissue morphology. Nat Commun 16, 4452 (2025). https://doi.org/10.1038/s41467-025-58989-8
Mots-clés: transcriptomique spatiale, morphologie tissulaire, pathologie numérique, prédiction de l'expression génique, IA pour l'imagerie