Clear Sky Science · fr
Catégorisation de 34 méthodes computationnelles pour détecter les gènes spatialement variables à partir de données de transcriptomique spatiale
Pourquoi l'emplacement compte pour nos gènes
Nos organismes sont constitués de cellules qui diffèrent non seulement par leurs fonctions, mais aussi par leur position au sein des tissus et des organes. Les nouvelles technologies de « transcriptomique spatiale » permettent désormais de lire quels gènes sont actifs tout en conservant l’adresse de chaque cellule sur une carte tissulaire. Cet article de synthèse explique comment les chercheurs détectent les gènes dont l’activité varie selon l’endroit — les fameux gènes spatialement variables — et pourquoi s’accorder sur les méthodes pour les repérer est essentiel pour comprendre le cancer, le fonctionnement du cerveau et de nombreuses autres maladies.
Des cellules éparses aux cartes vivantes
Les études traditionnelles en cellule unique mesurent l’activité génique de milliers de cellules individuelles mais perdent l’information sur leur origine spatiale. La transcriptomique spatiale comble cette lacune en mesurant l’activité des gènes directement sur des tranches de tissu. Chaque mesure est liée à un « point » sur la coupe, qui peut contenir une cellule ou plusieurs, selon la technologie. Les méthodes basées sur l’imagerie localisent quelques centaines de gènes choisis avec un très haut niveau de détail spatial, tandis que les plateformes par séquençage capturent presque tous les gènes mais à une résolution plus faible. Ensemble, ces approches transforment une section tissulaire en une carte colorée de l’activité génique capable de révéler des structures cachées, comme des couches du cerveau ou des régions à l’intérieur d’une tumeur.
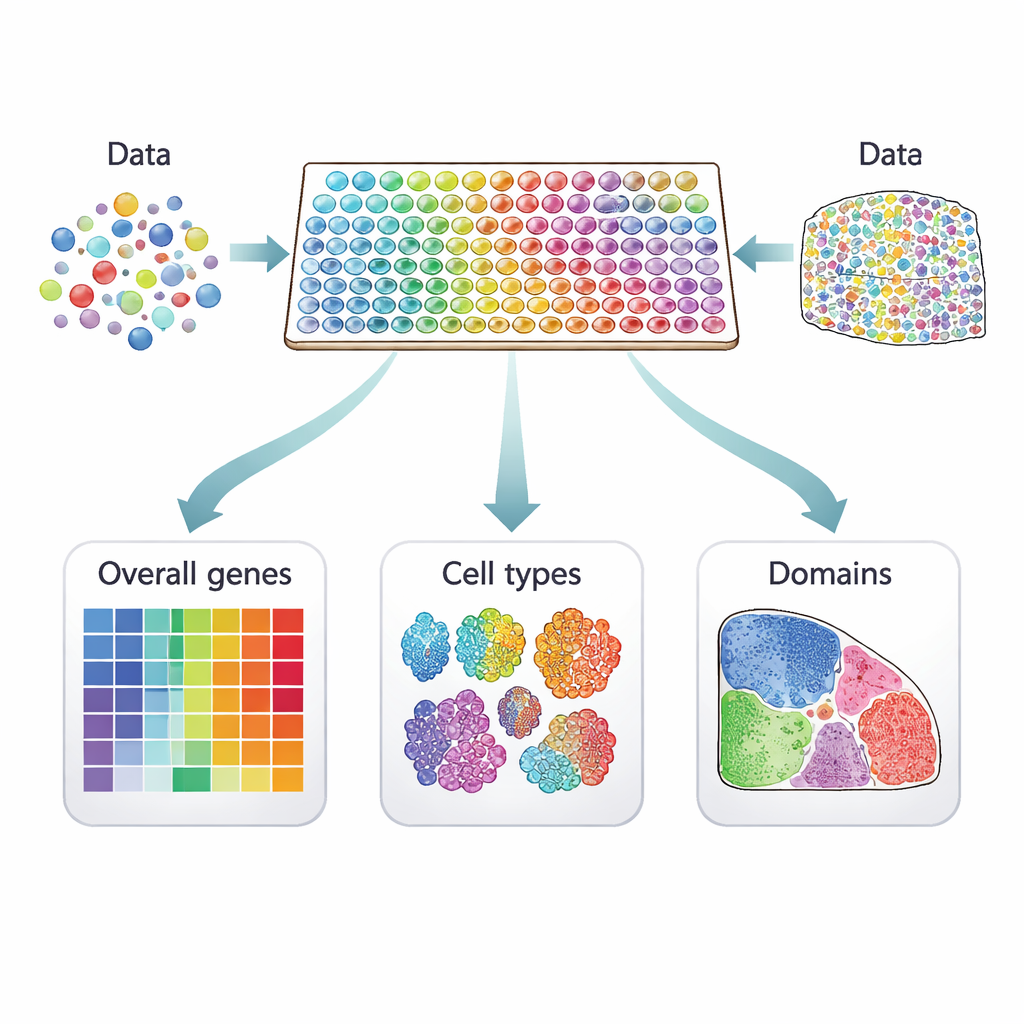
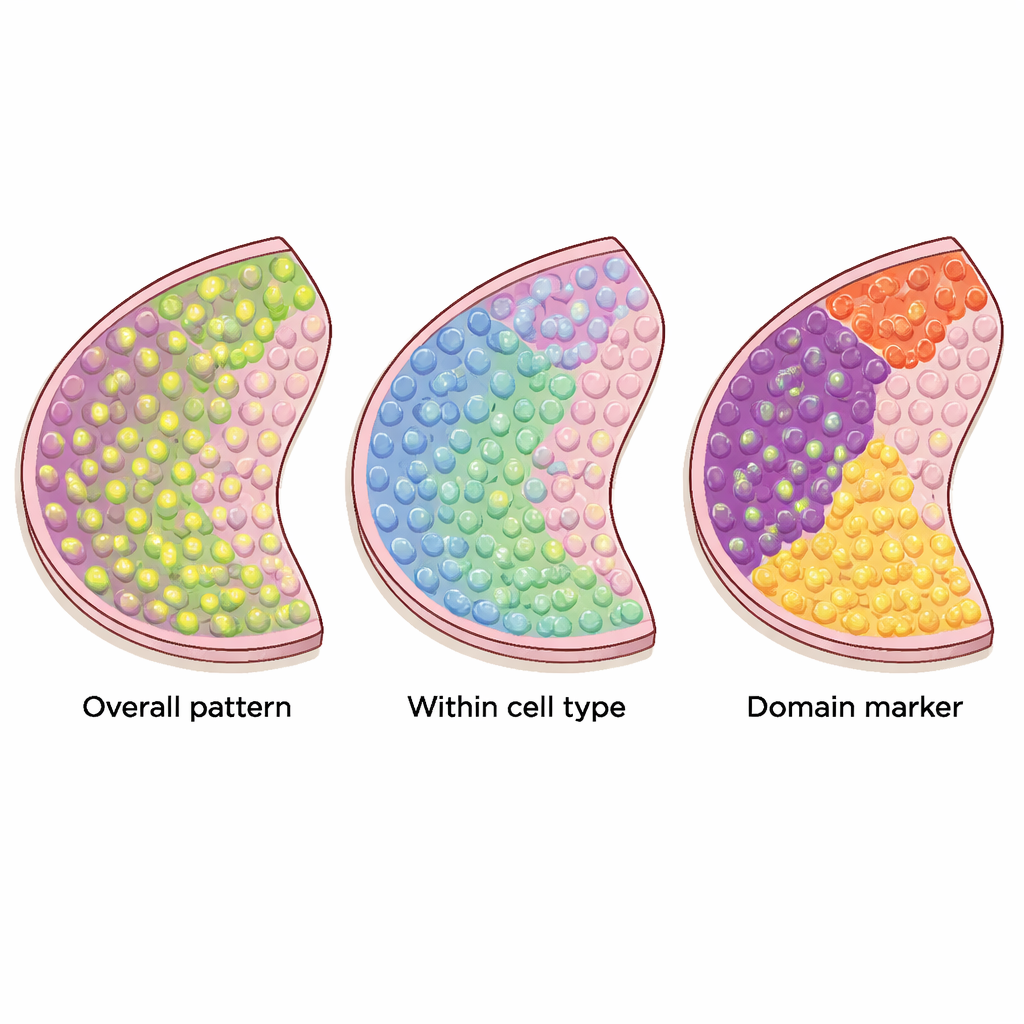
Trois types de gènes sensibles à la position
De nombreuses méthodes informatiques ont été proposées pour repérer les gènes présentant des motifs pertinents sur ces cartes tissulaires, mais elles ne cherchent pas toutes la même chose. Les auteurs classent 34 méthodes actuelles en trois catégories distinctes. Le premier groupe recherche les gènes spatialement variables « globaux », dont l’activité varie dans le tissu d’une manière non aléatoire — formant par exemple des bandes, des amas ou des gradients. Le deuxième groupe se concentre sur les gènes spatiaux « spécifiques à un type cellulaire » qui varient au sein d’un seul type de cellule, aidant à distinguer, par exemple, des sous‑types de neurones ou différents états des cellules immunitaires. Le troisième groupe cherche les gènes « marqueurs de domaines » fortement activés dans des régions ou des couches particulières, utiles comme étiquettes pour ces zones tissulaires.
Des outils différents pour des motifs différents
La revue explique le fonctionnement de ces méthodes en coulisses. Certaines traitent la coupe tissulaire comme des points dans un espace bidimensionnel régulier et utilisent des « noyaux » mathématiques pour détecter des motifs tels que taches ou ondes. D’autres relient d’abord les emplacements voisins en un réseau, ou graphe, puis évaluent si les fortes expressions ont tendance à se regrouper le long des arêtes de ce graphe. Certains outils reposent sur des tests statistiques formels aux taux d’erreur bien définis, tandis que d’autres se contentent de classer les gènes selon la netteté de leur motif. Les méthodes ciblant des motifs très spécifiques peuvent être puissantes lorsque les données correspondent à leurs attentes, mais elles risquent de passer à côté de formes plus irrégulières ou complexes, comme celles observées dans de nombreux cancers. Il existe aussi un compromis entre flexibilité et rapidité : certaines approches s’adaptent à des centaines de milliers de points, tandis que d’autres peinent avec des jeux de données très volumineux.
Ce que ces gènes peuvent révéler
Une fois identifiés, les gènes spatialement variables deviennent la matière première d’analyses biologiques plus approfondies. Les gènes spatiaux « globaux » sont souvent utilisés comme filtre initial pour réduire le nombre de gènes avant de regrouper les points en « domaines spatiaux » — des régions dont les cellules partagent des profils d’expression similaires. Ces domaines peuvent coïncider avec des structures tissulaires connues, suggérer de nouvelles sous‑régions ou mettre en lumière des voisinages cellulaires distincts, comme des fronts invasifs dans les tumeurs. Les gènes marqueurs de domaine aident ensuite à expliquer ce qui rend chaque région unique et peuvent être réutilisés pour annoter des structures similaires dans d’autres échantillons. Quant aux gènes spatiaux spécifiques à un type cellulaire, ils promettent une vue plus fine des variations d’un type cellulaire à travers un tissu, ce qui peut éclairer les interactions tumeur‑immunité ou les circuits spécialisés dans le cerveau.
Défis et perspectives
Les auteurs soulignent qu’aucune méthode n’est universellement la meilleure et que comparer les outils équitablement exige de réfléchir soigneusement au type de gène spatial que chaque méthode vise réellement à détecter. Ils appellent à de meilleurs jeux de référence réalistes, à des standards statistiques plus clairs pour éviter les fausses découvertes, et à de nouvelles approches qui prennent en compte les différences entre technologies et types de tissus. Pour les non‑spécialistes, le message clé est que les gènes spatialement variables transforment des listes plates de gènes en cartes vivantes, reliant l’activité moléculaire à la structure tissulaire. Des méthodes robustes pour détecter et interpréter ces gènes seront centrales pour faire de la transcriptomique spatiale un outil concret pour étudier le développement, le fonctionnement cérébral et les maladies.
Citation: Yan, G., Hua, S.H. & Li, J.J. Categorization of 34 computational methods to detect spatially variable genes from spatially resolved transcriptomics data. Nat Commun 16, 1141 (2025). https://doi.org/10.1038/s41467-025-56080-w
Mots-clés: transcriptomique spatiale, gènes spatialement variables, motifs d'expression génique, microenvironnements tissulaires, génomique computationnelle