Clear Sky Science · fr
L’inhibition de RRM1 sensibilise l’adénocarcinome pulmonaire au traitement par décitabine
Transformer un médicament tiède en un allié plus puissant
Le cancer du poumon reste l’un des cancers les plus meurtriers, et de nombreux patients finissent par manquer d’options thérapeutiques efficaces. Les cliniciens espèrent depuis longtemps que des médicaments qui reprogrammèrent légèrement l’ADN des cellules cancéreuses, plutôt que de simplement empoisonner les cellules en division, pourraient aider. Un de ces médicaments, la décitabine, fonctionne bien dans les cancers du sang mais a déçu dans les tumeurs solides comme le cancer du poumon. Cette étude pose une question simple et pragmatique aux implications majeures : existe‑t‑il un moyen de rendre enfin les tumeurs pulmonaires réceptives à la décitabine, en utilisant des outils que nous comprenons déjà ?
Pourquoi un médicament éprouvé échoue dans les tumeurs solides
La décitabine est un analogue d’un des éléments constitutifs de l’ADN. Lorsqu’elle s’insère dans l’ADN d’une cellule au moment de la réplication, elle peut effacer des marques chimiques anormales qui silencient des gènes protecteurs, y compris des suppresseurs de tumeurs et des gènes impliqués dans l’immunité. Dans les leucémies, cela aide à réorienter les cellules vers un état plus sain. Dans les tumeurs pulmonaires, en revanche, le médicament a à peine d’effet. Les auteurs ont suspecté que le problème ne venait pas de l’action de la décitabine, mais de la faible quantité qui parvient effectivement à s’introduire dans l’ADN des cellules de tumeur solide. En mesurant de toutes petites quantités de médicament incorporées à l’ADN dans de nombreuses lignées cellulaires cancéreuses, ils ont confirmé que les cellules qui incorporaient davantage de décitabine étaient beaucoup plus faciles à tuer par le médicament.
Un gardien cellulaire qui bloque le médicament
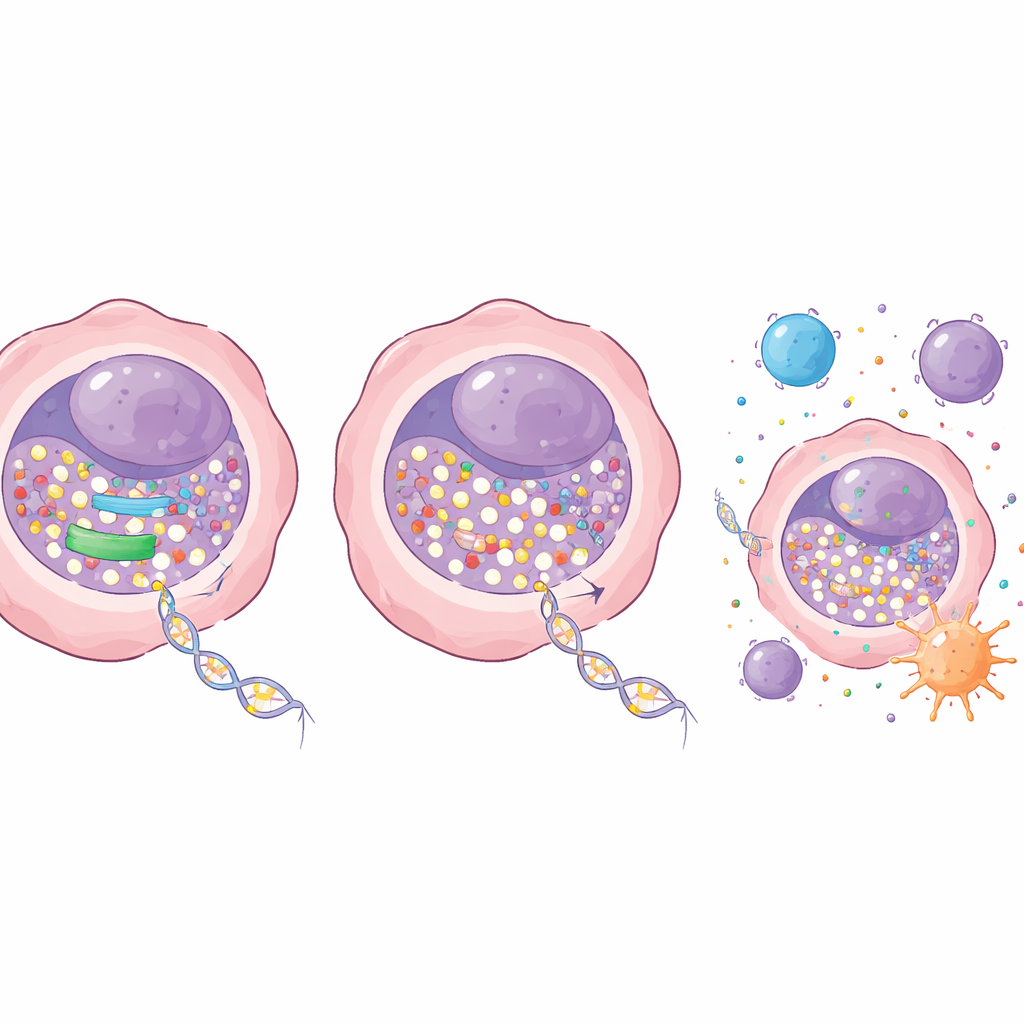
Pour identifier ce qui limite l’entrée du médicament dans l’ADN, les chercheurs ont examiné les gènes impliqués dans la gestion des nucléosides — les matériaux bruts de l’ADN. Une enzyme, nommée RRM1, a émergé. RRM1 fait partie d’une machinerie qui convertit les éléments ordinaires en formes actives utilisées pour synthétiser l’ADN. Dans l’adénocarcinome pulmonaire, cette enzyme était anormalement abondante dans les tumeurs comparées au tissu pulmonaire normal, et les patients présentant des niveaux plus faibles de RRM1 avaient tendance à vivre plus longtemps. Dans un panel de lignées cellulaires cancéreuses, des niveaux plus élevés de RRM1 allaient de pair avec une incorporation plus faible de décitabine, suggérant fortement que cette enzyme agit comme un gardien qui écarte le médicament.
Désarmer le gardien pour aider le médicament à agir
L’équipe a ensuite testé ce qui se passe lorsqu’on désactive partiellement RRM1. À l’aide d’outils génétiques, ils ont diminué l’expression de RRM1 dans des cellules de cancer pulmonaire sans tuer les cellules directement. Pris isolément, cette réduction n’avait qu’un effet modéré sur la croissance. Mais combinée à de faibles doses de décitabine, l’impact fut spectaculaire : les colonies de cellules cancéreuses pulmonaires se sont nettement réduites en culture, et les tumeurs ont poussé beaucoup plus lentement chez la souris. Il est important de noter que ces doses efficaces étaient bien tolérées, sans atteinte évidente de la fonction sanguine, hépatique ou rénale chez les animaux. Au niveau moléculaire, le blocage de RRM1 permettait à plus de décitabine d’être incorporée à l’ADN, entraînant une perte plus marquée de l’enzyme ajoutant des méthyles, DNMT1, et une baisse plus importante de la méthylation globale de l’ADN. Cela a, à son tour, réactivé des gènes suppresseurs de tumeurs qui avaient été mis hors fonction.
Activer l’alarme immunitaire à l’intérieur des tumeurs
Au‑delà du ralentissement de la division cellulaire, la thérapie combinée a modifié la manière dont les cellules cancéreuses interagissent avec le système immunitaire. L’excès de décitabine dans l’ADN a augmenté les signaux de dommage à l’ADN à l’intérieur des cellules et les a poussées vers la mort programmée. Parallèlement, il a stimulé l’activité d’un système d’alarme interne centré sur la voie STING, qui détecte l’ADN déplacé et déclenche des réponses immunitaires de type antiviral. Lorsque RRM1 était bloquée, la décitabine activait plus fortement cette voie et ses gènes en aval, y compris ceux qui recrutent et stimulent les cellules immunitaires. Dans des modèles murins de cancer du poumon avec un système immunitaire intact, la combinaison de décitabine et d’un inhibiteur de RRM1 a permis de mieux contrôler les tumeurs que chaque traitement pris séparément, sans toxicité évidente supplémentaire. Les auteurs ont également constaté que cette stratégie d’inhibition de l’enzyme potentialise spécifiquement la décitabine, et peut en réalité être moins efficace voire nuisible avec un médicament apparenté, l’azacitidine, soulignant la nécessité d’associer les bons partenaires.
Ce que cela pourrait signifier pour les patients
En somme, ce travail dresse un tableau clair : une enzyme de synthèse d’éléments d’ADN trop active dans les tumeurs pulmonaires limite la quantité de décitabine atteignant sa cible. En bloquant partiellement cette enzyme, les cellules cancéreuses sont contraintes d’utiliser davantage le médicament à la place de leurs composants habituels. Ce basculement permet à de faibles doses de décitabine de désilencer plus efficacement des gènes protecteurs, d’endommager l’ADN des cellules cancéreuses et d’éveiller les défenses immunitaires, tout en restant tolérable dans les modèles animaux. Pour les patients, cela suggère une voie réaliste : réutiliser ou affiner des inhibiteurs de RRM1, en association avec de faibles doses de décitabine et éventuellement des immunothérapies modernes, pour transformer un médicament autrefois décevant en un élément utile du traitement du cancer du poumon.
Citation: Jiang, N., Liu, J., Vaghasia, A. et al. RRM1 inhibition sensitizes lung adenocarcinoma to decitabine treatment. Cell Death Dis 17, 275 (2026). https://doi.org/10.1038/s41419-026-08522-6
Mots-clés: adénocarcinome pulmonaire, décitabine, méthylation de l’ADN, ribonucléotide réductase, immunothérapie du cancer