Clear Sky Science · fr
Des variants perte de fonction d’HPDL altèrent le développement cortical humain via des modifications de la fonction mitochondriale
Pourquoi les petits moteurs cellulaires comptent pour les cerveaux en croissance
La plupart des gens pensent surtout à la génétique et au câblage lorsqu’ils imaginent le développement du cerveau. Cette étude montre qu’un autre facteur, souvent négligé — les petites centrales énergétiques à l’intérieur de nos cellules appelées mitochondries — peut aussi influencer la formation du cerveau. En étudiant des troubles moteurs pédiatriques rares liés au gène HPDL, les auteurs révèlent comment une production d’énergie défectueuse peut réduire le cortex en développement, la région cérébrale cruciale pour le mouvement, la pensée et le comportement.
Un trouble moteur rare comme fenêtre sur la croissance cérébrale
Certain·e·s enfants porteurs de variants du gène HPDL développent une paraplégie spastique héréditaire, une affection provoquant raideur et faiblesse des jambes, accompagnée de crises, d’un retard du développement et, dans les cas sévères, d’un cerveau plus petit que la normale (microcéphalie). Bien que l’on sache que la protéine HPDL se situe dans les mitochondries, sa fonction exacte — et la raison pour laquelle sa perte endommage le cerveau — restait incertaine. Les chercheurs ont utilisé plusieurs modèles cellulaires humains, incluant des cellules tumorales de type neuronal et des cellules cérébrales dérivées de prélèvements cutanés de patients, pour tester si HPDL est nécessaire au développement cérébral normal et à la santé mitochondriale.
Que se passe-t-il quand HPDL est désactivé
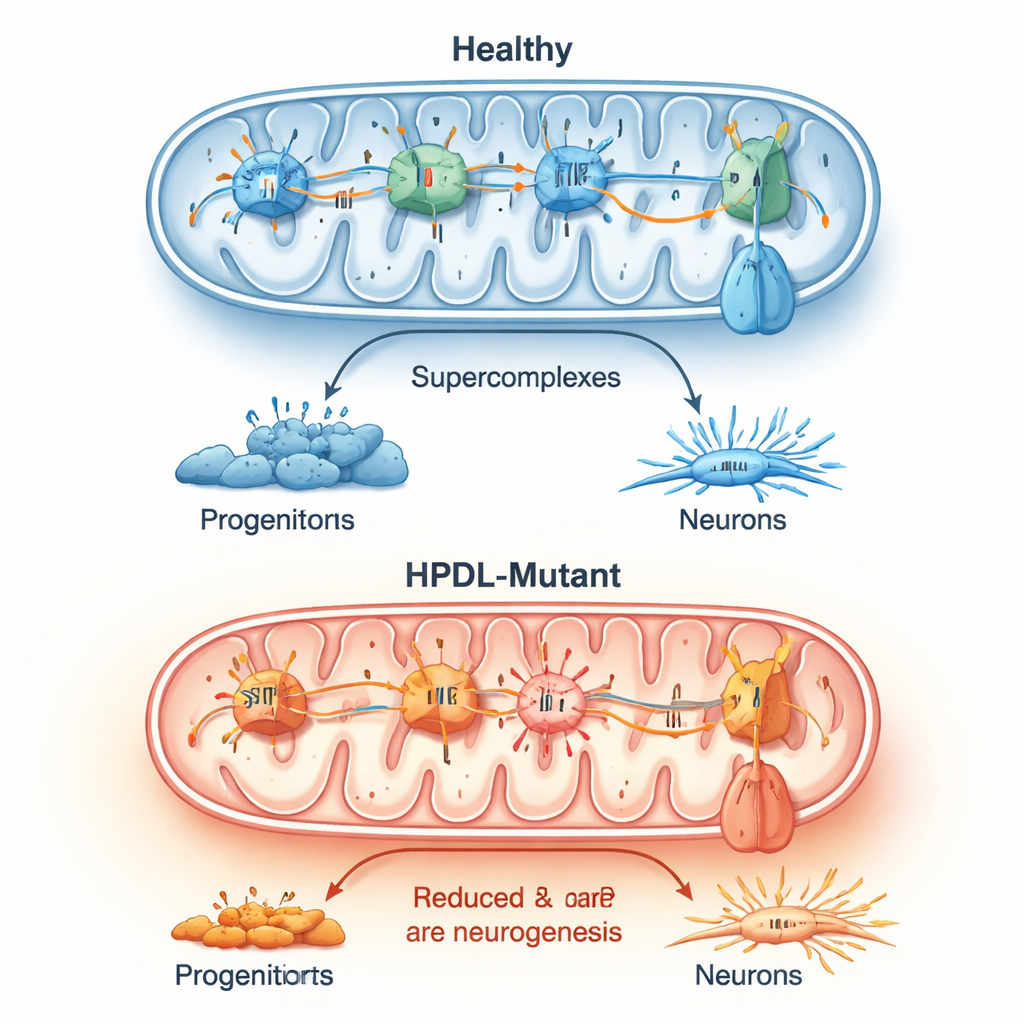
Tout d’abord, l’équipe a inactivé HPDL dans une lignée humaine de neuroblastome à l’aide de l’édition génomique CRISPR. Sans HPDL, ces cellules ont perdu la protéine pleine longueur et ont montré des problèmes mitochondriaux évidents. Les grandes assemblées de protéines de la chaîne respiratoire qui travaillent normalement ensemble pour générer de l’énergie étaient perturbées, et des composants clés impliqués dans l’utilisation de l’oxygène étaient réduits. Les cellules consommaient moins d’oxygène, produisaient moins de respiration liée à l’énergie et généraient davantage d’espèces réactives de l’oxygène — des sous-produits nocifs souvent qualifiés de « stress oxydatif ». Pourtant le nombre total de mitochondries n’a pas diminué, et les niveaux de coenzyme Q10, une molécule essentielle au transfert d’énergie, étaient en réalité plus élevés, ce qui suggère un défaut qualitatif — et pas seulement quantitatif — de la fonction mitochondriale.
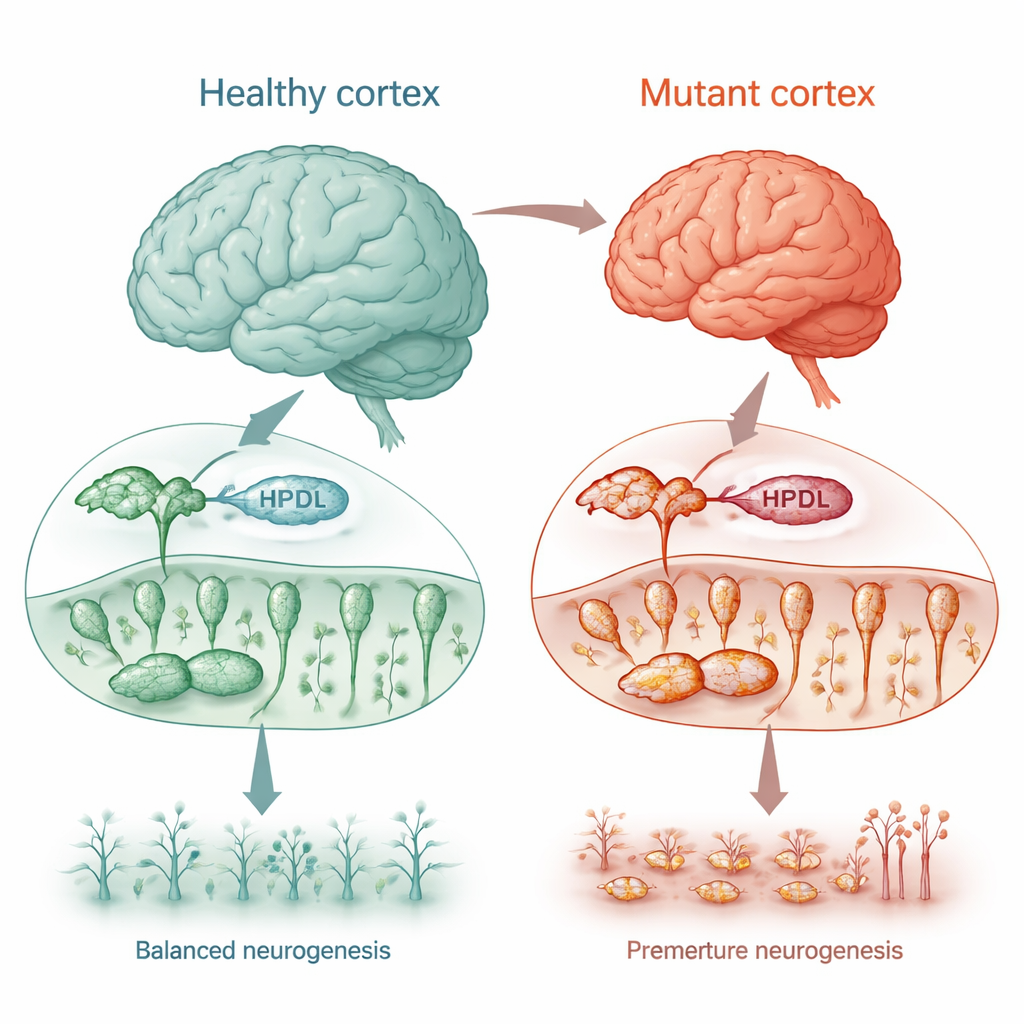
Du tissu cérébral en boîte révèle une surproduction précoce de neurones
Pour voir comment la perte d’HPDL affecte le véritable développement cérébral humain, les chercheurs ont reprogrammé des cellules de peau de quatre enfants concernés en cellules souches pluripotentes induites, puis les ont orientées vers des cellules corticales et des « mini-cerveaux » tridimensionnels (organoïdes). Tôt dans le développement, à un stade où la plupart des cellules devraient encore se diviser en tant que progéniteurs neuronaux, les cultures mutantes pour HPDL contenaient déjà davantage de neurones matures et moins de progéniteurs. Les profils d’activité génique ont confirmé cela : les voies qui favorisent la formation neuronale étaient activées trop tôt, tandis que celles maintenant les cellules dans un état prolifératif étaient diminuées. Dans les organoïdes, ce passage prématuré des éléments de construction aux neurones achevés a conduit à des structures de type cérébral beaucoup plus petites, rappelant la microcéphalie observée chez les enfants les plus atteints.
Des centrales énergétiques défaillantes et des cellules en détresse
Une inspection plus fine a montré que les cellules cérébrales mutantes pour HPDL présentaient une phosphorylation oxydative altérée — la principale voie par laquelle les mitochondries produisent de l’énergie. Des colorations enzymatiques ont révélé une activité plus faible d’un complexe mitochondrial clé, tandis que d’autres mesures ont montré une charge électrique modifiée à travers la membrane mitochondriale. Dans de nombreuses cellules mutantes, une enzyme cruciale qui produit normalement de l’ATP semblait fonctionner à l’inverse pour maintenir cette charge membranaire, signe d’un profond désordre métabolique. Chez l’ensemble des lignées de patients, les espèces réactives de l’oxygène étaient systématiquement élevées, et les grandes assemblées normales de protéines de la chaîne respiratoire étaient moins bien formées. Ces changements mitochondriaux suivaient étroitement le moment et le degré de la production neuronale prématurée.
Tester des moyens d’atténuer le stress
Comme le stress oxydatif et l’altération de la chimie du coenzyme Q10 semblaient centraux, l’équipe a testé si des traitements ciblant ces problèmes pouvaient ralentir l’entrée hâtive en neurogenèse. Ils ont exposé des cultures corticales précoces à deux antioxydants et au 4‑hydroxybenzoate, une petite molécule liée à la synthèse du coenzyme Q10. Dans plusieurs lignées dérivées de patients, ces composés ont partiellement réduit la neurogenèse prématurée, mais la réponse dépendait de la mutation HPDL précise. Certaines lignées ont surtout répondu aux antioxydants, d’autres au précurseur du coenzyme Q10, et l’une n’a pas du tout répondu. Ce schéma spécifique aux mutations suggère que des stratégies thérapeutiques personnalisées pourraient être nécessaires pour les troubles liés à HPDL.
Ce que cela signifie pour les enfants et les thérapies futures
En termes simples, cette étude montre que HPDL agit comme un gardien des éléments constitutifs du cerveau pendant le développement précoce. Lorsque HPDL fait défaut, les mitochondries deviennent inefficaces et excessivement stressées, poussant les cellules progénitrices à devenir des neurones trop tôt. Le réservoir de cellules en division s’épuise, le cortex ne peut atteindre sa taille maximale et les schémas de câblage sont altérés, contribuant aux troubles moteurs et à d’autres symptômes. Le secours partiel observé avec des antioxydants et des composés liés au coenzyme Q10 suggère que rééquilibrer l’énergie cellulaire et le stress oxydatif pourrait un jour aider les enfants porteurs de mutations HPDL, et peut-être d’autres atteints de maladies cérébrales d’origine mitochondriale.
Citation: Baggiani, M., Desbats, M.A., Naef, V. et al. Loss of function variants in HPDL impair human cortical development via alterations of mitochondrial function. Cell Death Dis 17, 237 (2026). https://doi.org/10.1038/s41419-026-08476-9
Mots-clés: HPDL, mitochondries, développement cortical, microcéphalie, stress oxydatif