Clear Sky Science · fr
L’entretien aberrant du facteur de transcription du développement PAX6 favorise la mort neuronale via la signalisation JNK3
Pourquoi cette recherche est importante pour la vision
Le glaucome est une cause majeure de cécité permanente, principalement parce que les cellules nerveuses qui transmettent l’information visuelle de l’œil au cerveau meurent lentement. De nombreux traitements abaissent la pression intraoculaire, mais la perte de vision peut se poursuivre même lorsque la pression est bien contrôlée. Cette étude pose une question plus profonde : qu’est-ce qui pousse ces cellules ganglionnaires rétiniennes à engager la mort lorsqu’elles sont en stress, et peut-on éteindre cette décision au niveau du contrôle génétique à l’intérieur du noyau cellulaire ?
Une rétine stressée sous attaque
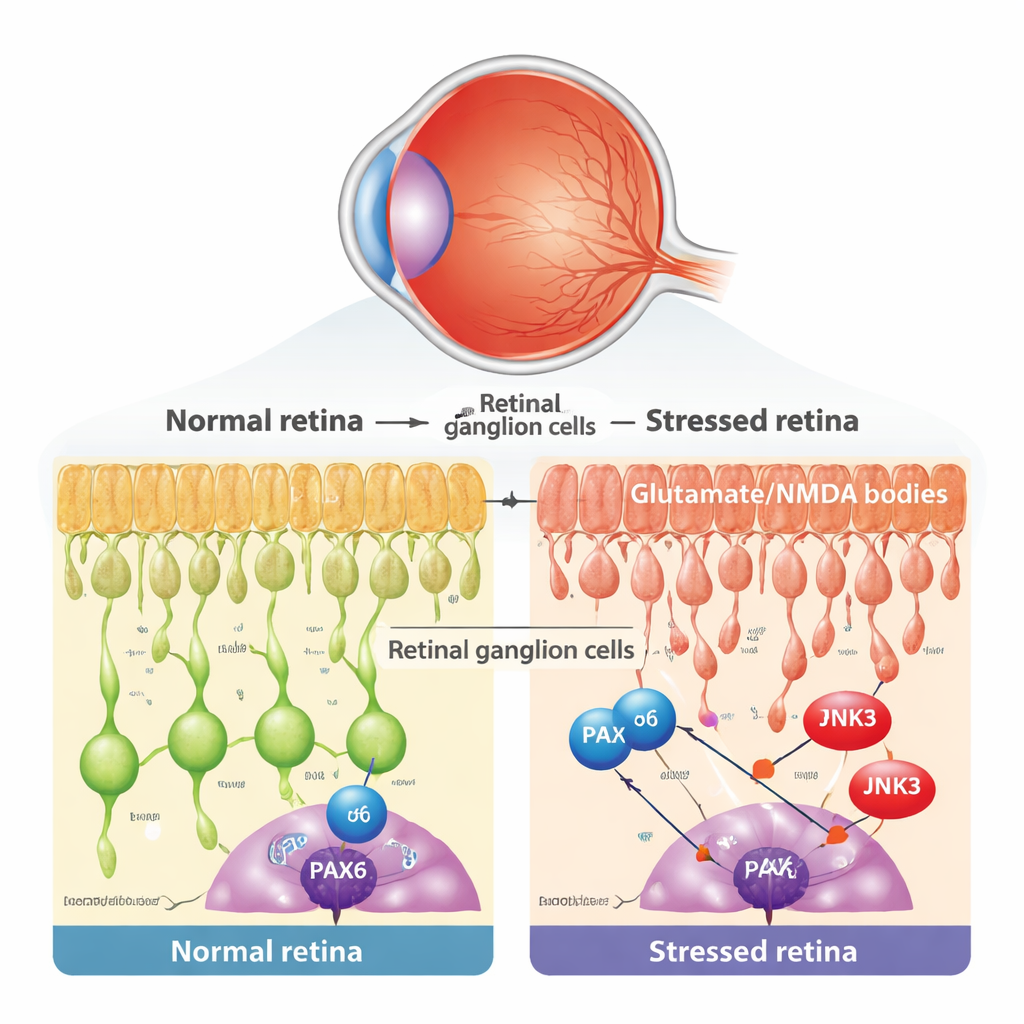
Au cœur du glaucome et des maladies oculaires apparentées se trouve la perte progressive des cellules ganglionnaires rétiniennes (CGR), les neurones effecteurs de la rétine. Ces cellules sont vulnérables à de nombreux types de stress, y compris des niveaux toxiques du neurotransmetteur glutamate, qui suractive les récepteurs NMDA et déclenche une surcharge calcique délétère. Les chercheurs ont utilisé un modèle murin bien établi dans lequel une petite quantité de NMDA est injectée dans l’œil, blessant sélectivement les CGR tout en laissant les autres couches rétiniennes en grande partie intactes. Ils ont confirmé que ce traitement ne modifiait pas la pression oculaire, mais provoquait des signes typiques d’apoptose des CGR, tels que la libération de cytochrome c par les mitochondries et l’apparition de noyaux positifs au TUNEL.
Un gène du développement qui refuse de prendre sa retraite
Pendant le développement précoce, un régulateur génique appelé PAX6 joue le rôle d’architecte maître de l’œil, guidant la naissance et le câblage des différentes cellules rétiniennes. La sagesse conventionnelle veut que ces programmes de développement s’éteignent en grande partie à l’âge adulte. En réanalysant des données de séquençage d’ARN monocellulaire provenant de rétines de souris et d’humains, l’équipe a trouvé que PAX6 est en réalité fortement et sélectivement maintenu dans les CGR matures et certains interneurones. Par des marquages microscopiques, ils ont montré que dans la couche où résident les CGR, PAX6 est majoritairement présent dans les cellules ganglionnaires plutôt que dans les cellules amacrines voisines. Cela a ouvert une possibilité intrigante : chez l’adulte malade, un ancien programme développemental pourrait être réutilisé et transformé en moteur de dégénérescence.
Du gardien à l’exécuteur : PAX6 change de rôle
Pour tester si PAX6 aide les CGR à survivre ou à mourir sous stress, les scientifiques ont employé une approche de type thérapie génique. Ils ont délivré un vecteur viral transportant un petit ARN qui réduit spécifiquement l’expression de PAX6 dans la rétine, puis ont exposé les yeux au NMDA. Par rapport aux yeux traités-témoins, les rétines appauvries en PAX6 présentaient beaucoup moins de CGR apoptotiques et beaucoup moins de dommages mitochondriaux, indiquant que PAX6 est nécessaire pour la mort cellulaire complète dans ce modèle. Le séquençage global de l’ARN a révélé que de nombreux gènes pro-mort, en particulier ceux impliqués dans les dommages mitochondriaux et l’activation des caspases, étaient fortement induits par le NMDA chez des souris normales mais atténués lorsque PAX6 était silencé. Autrement dit, PAX6 contribue à activer un réseau de gènes qui poussent les CGR au-delà du seuil de survie.
La kinase de stress qui déclenche l’interrupteur PAX6
Comment le stress active-t-il PAX6 sans en augmenter la quantité ? L’équipe s’est concentrée sur JNK3, une enzyme sensible au stress présente principalement dans les neurones. Lors de la lésion par NMDA, JNK3 migrait vers le noyau des CGR et s’associait physiquement à PAX6. Des expériences biochimiques « en éprouvette » avec des protéines purifiées ont montré que JNK3 peut directement ajouter des groupements phosphate sur PAX6, et cette réaction était bloquée par un inhibiteur de JNK. Chez des souris dépourvues du gène Jnk3, le NMDA ne produisait plus le même profil de phosphorylation de PAX6. Le cartographiage de la chromatine (ChIP-seq) et des tests ciblés de liaison à l’ADN ont révélé que, sous stress, PAX6 phosphorylé, conjointement avec JNK3, se lie plus fortement aux régions régulatrices de gènes pro-apoptotiques clés comme Bax et Gadd45a, augmentant leur activité. Lorsque PAX6 était réduit ou que JNK3 était supprimé génétiquement, cette liaison et l’activation correspondante des gènes pro-mort étaient fortement atténuées.
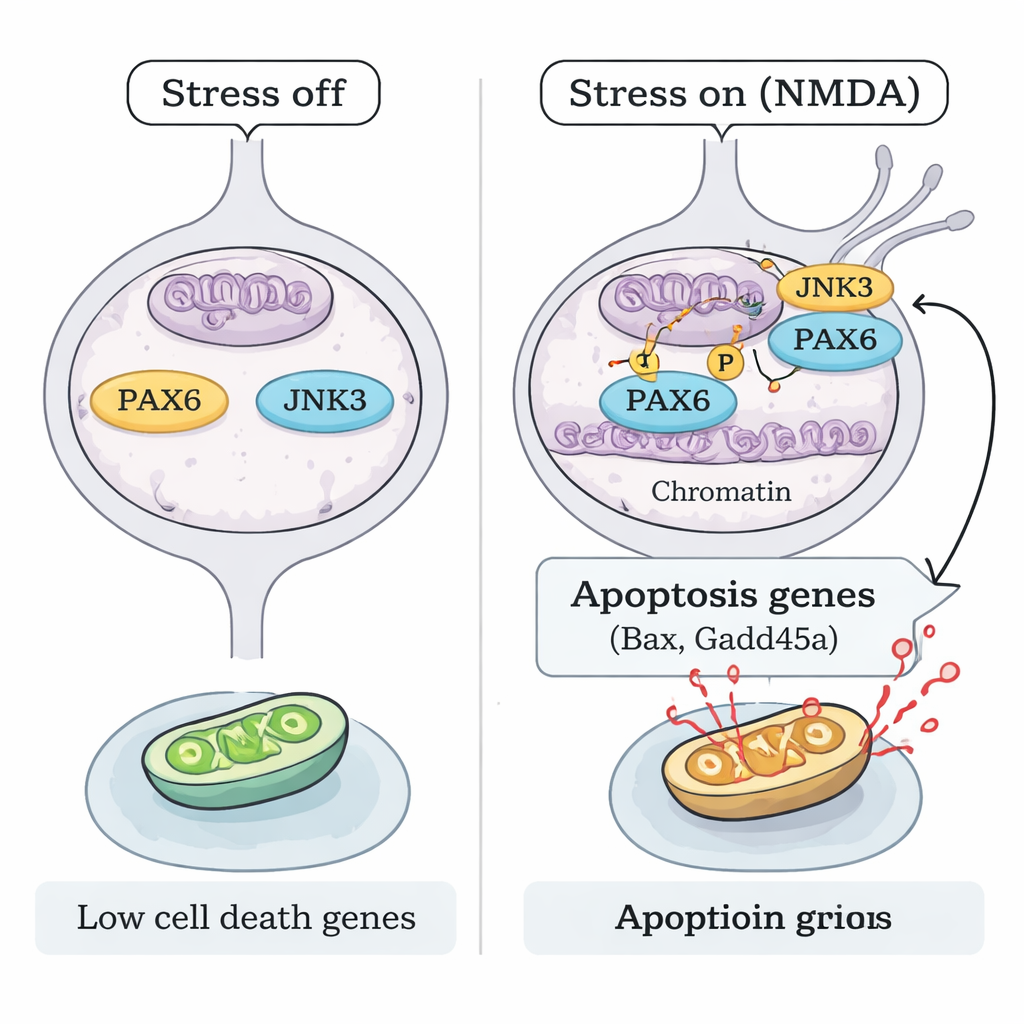
Éteindre le programme de mort pour protéger la vision
Enfin, les chercheurs ont testé si bloquer cet axe JNK3–PAX6 suffit à protéger les cellules essentielles à la vision. Tant chez les souris avec réduction de PAX6 que chez celles déficientes en JNK3, les CGR ont été significativement préservées après exposition au NMDA, avec moins de cellules mortes et une structure rétinienne plus saine. Cela aboutit à un modèle mécanistique clair : sous stress excitotoxique, JNK3 phosphoryle le PAX6 exprimé de manière persistante, le convertissant d’un constructeur développemental en un activateur puissant d’un programme génétique de mort cellulaire dans les CGR adultes. Interrompre ce lien — en silencant PAX6 ou en désactivant JNK3 — permet de maintenir en vie un grand nombre de ces neurones. Pour les patients, ce travail suggère que les futures thérapies du glaucome pourraient aller au-delà de la réduction de la pression oculaire et cibler directement les interrupteurs génétiques qui décident de la survie ou de la mort des neurones rétiniens.
Citation: Kim, JY., An, MJ., Kim, J. et al. Aberrant maintenance of developmental transcription factor PAX6 promotes neuronal cell death via JNK3 signaling. Cell Death Dis 17, 161 (2026). https://doi.org/10.1038/s41419-026-08417-6
Mots-clés: glaucome, cellules ganglionnaires rétiniennes, PAX6, JNK3, neurodégénérescence