Clear Sky Science · fr
Renforcer l’activité de KLF15 dans les cardiomyocytes : une nouvelle approche pour prévenir le reprogrammation pathologique et la fibrose via dCas9VPR dépourvu de nucléase
Reprogrammer le cœur en insuffisance
L’insuffisance cardiaque touche des millions de personnes, se développant souvent lentement après des années d’hypertension ou de maladie valvulaire. Dans ces conditions, les cellules musculaires du cœur augmentent non seulement de taille mais réactivent aussi un programme génétique « fœtal » et le cœur se remplit de tissu cicatriciel. Cette étude explore une nouvelle façon de pousser la machinerie de contrôle génétique du cœur vers un état sain — sans couper l’ADN — en augmentant délicatement l’activité d’un régulateur protecteur nommé KLF15 dans les cardiomyocytes.
Quand les cellules cardiaques perdent leur identité
Dans un cœur adulte sain, les cardiomyocytes — les cellules musculaires cardiaques — oxydent efficacement les lipides pour produire de l’énergie et maintiennent un profil d’expression génique stable. En utilisant le séquençage d’ARN unicellulaire chez des souris soumises à une surcharge de pression chronique, les chercheurs ont cartographié comment chaque cardiomyocyte évolue lorsque le cœur passe d’un fonctionnement normal à une hypertrophie puis à l’insuffisance. Ils ont constaté qu’un facteur de transcription nommé KLF15, qui régule normalement l’équilibre entre métabolisme et croissance, présentait la plus forte variation d’activité dans les cellules malades. À mesure que le stress augmentait, les niveaux de KLF15 chutaient et sa capacité à réprimer les gènes fœtaux et liés au stress diminuait. Des baisses similaires de KLF15 ont été observées dans des cœurs humains provenant de patients atteints de cardiomyopathie dilatée et hypertrophique, indiquant que cette perturbation est conservée entre les espèces.
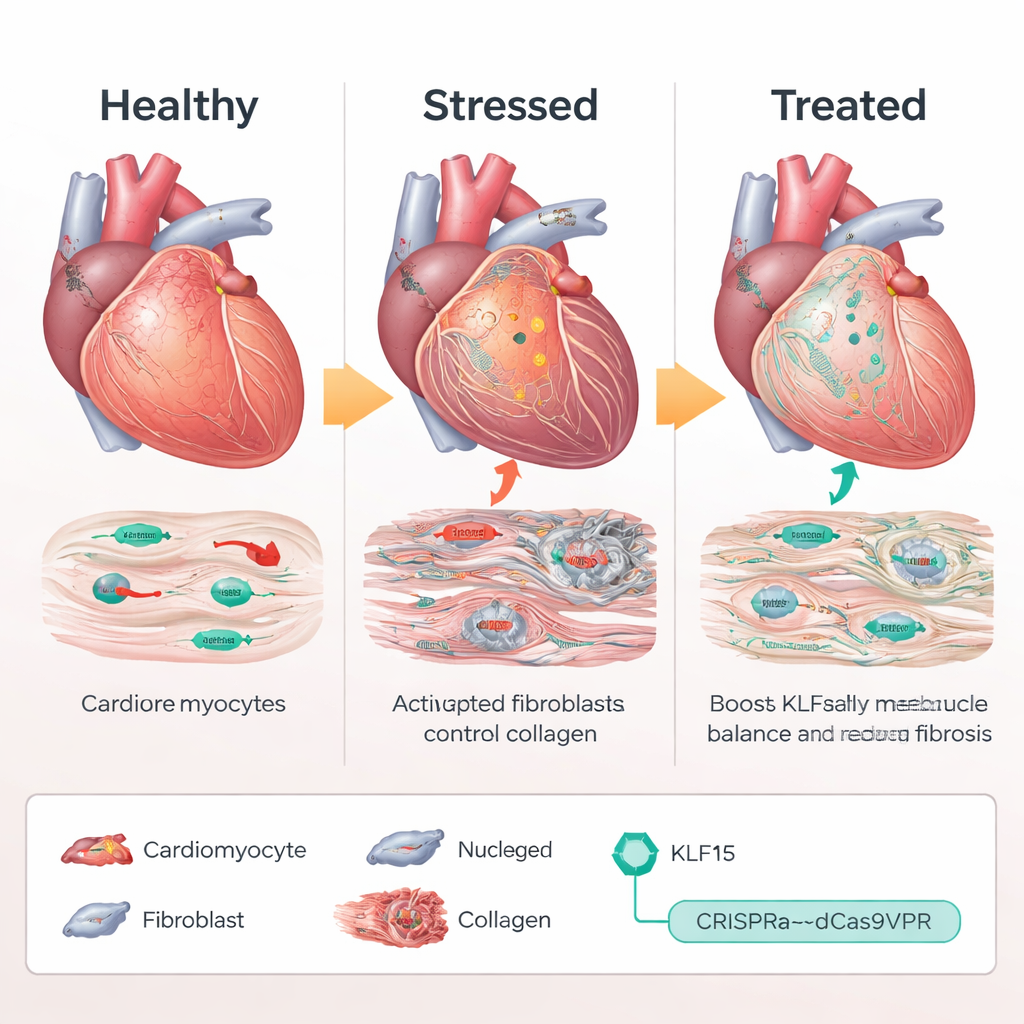
Utiliser CRISPR comme un bouton de volume, pas comme des ciseaux
Plutôt que d’ajouter une copie supplémentaire du gène KLF15 ou de couper l’ADN, l’équipe a utilisé un système d’« activation » basé sur CRISPR, nommé dCas9VPR, qui se lie à proximité du gène Klf15 endogène et augmente son expression. Chez des souris conçues pour exprimer cet activateur CRISPR uniquement dans les cardiomyocytes, les scientifiques ont délivré des ARN guides via un adénovirus associé (AAV9) ciblant le promoteur de Klf15. Sous surcharge de pression chronique, les souris recevant des guides activant Klf15 ont maintenu des niveaux proches de la normale de Klf15. Leurs cardiomyocytes sont restés de plus petite taille, la fonction de pompage a décliné moins fortement et la survie s’est améliorée comparée aux animaux témoins. Au niveau moléculaire, les gènes liés au stress et au profil fœtal se sont tus, tandis que les gènes du métabolisme et de la gestion du calcium ont repris de l’activité, indiquant que le programme transcriptionnel délétère avait été largement réinitialisé.
Freiner la formation de cicatrices par le dialogue intercellulaire
L’insuffisance cardiaque est portée non seulement par des cardiomyocytes malades mais aussi par des fibroblastes, cellules de soutien qui produisent du collagène et forment un tissu cicatriciel rigide. Des analyses unicellulaires et des images tissulaires ont montré que la restauration de Klf15 dans les cardiomyocytes réduisait l’activation des fibroblastes et la fibrose globale, bien que la thérapie génique n’ait jamais ciblé les fibroblastes directement. L’équipe a rattaché cet effet à une protéine sécrétée appelée AZGP1. Lorsque Klf15 était augmenté dans les cardiomyocytes, la production et la libération d’AZGP1 augmentaient. Tant dans des cœurs de souris que dans des tissus cardiaques dérivés de cellules souches humaines, des niveaux élevés d’AZGP1 ont atténué la voie TGF-β / SMAD dans les fibroblastes — un moteur clé de la cicatrisation — diminuant l’expression de marqueurs comme α-SMA et POSTN. Il est important de noter que la surexpression d’AZGP1 uniquement dans les cardiomyocytes n’a pas reprogrammé ces cellules musculaires, montrant que KLF15 protège principalement les cardiomyocytes de manière directe et utilise AZGP1 comme messager pour réfréner les fibroblastes.
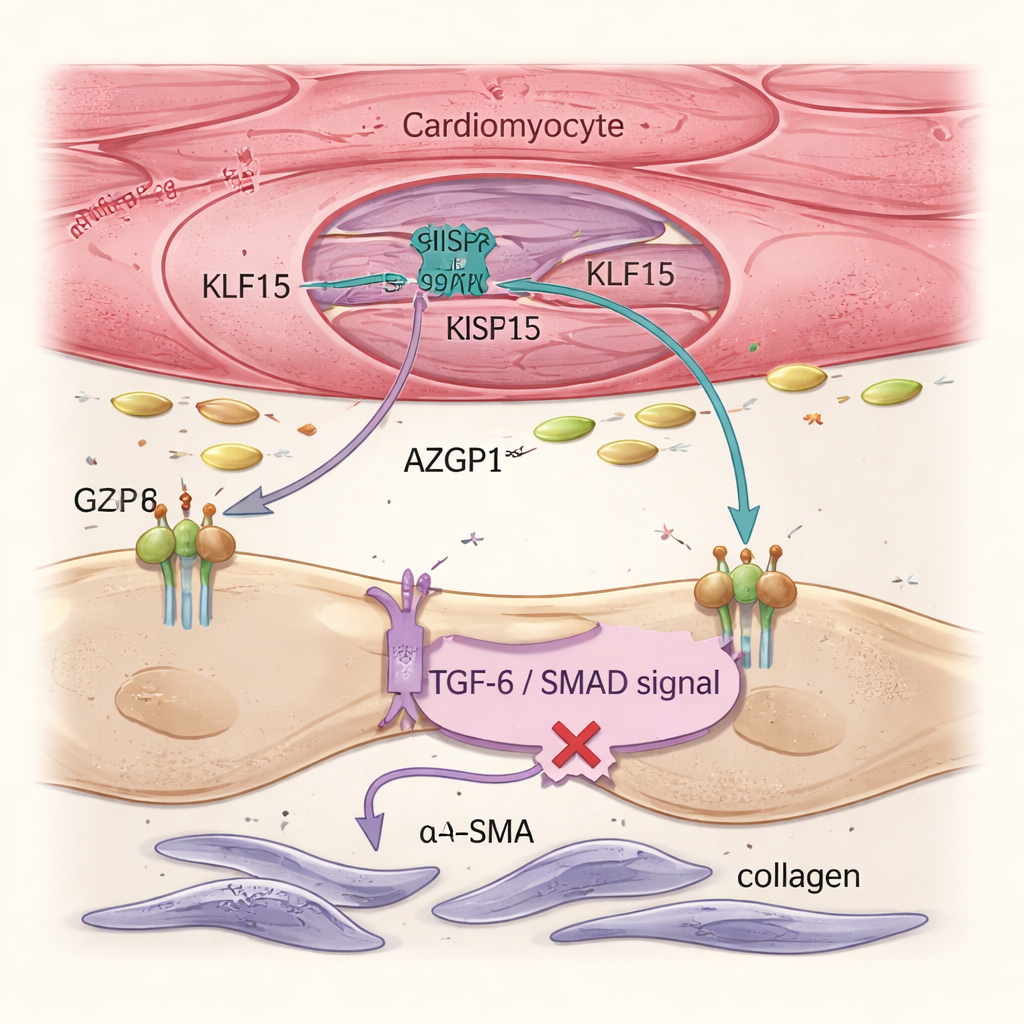
Des modèles tissulaires humains confirment le circuit protecteur
Pour vérifier si ces mécanismes s’appliquent aux cellules humaines, les chercheurs ont utilisé des cardiomyocytes dérivés de cellules souches pluripotentes induites cultivés dans des tissus cardiaques tridimensionnels. Lorsqu’ils ont été soumis à une charge mécanique imitant l’hypertension, ces tissus perdaient KLF15, activaient des gènes de stress et fœtaux, se rigidifiaient et leurs contractions s’affaiblissaient — recréant les traits de la maladie. La restauration de KLF15 par CRISPRa a empêché ce déclin, préservé la génération de force et recentré l’expression génique vers un métabolisme et une structure matures. Des expériences détaillées ont montré que la TGF-β1, un signal pro‑fibrotique bien connu, réduit KLF15 dans les cardiomyocytes humains via la voie SMAD2/3, ce qui aide à expliquer comment le stress chronique conduit à un remodelage maladaptatif. Enfin, l’équipe a conçu un système CRISPRa compact, « mini », basé sur une variante de Cas9 plus petite pouvant tenir dans un seul vecteur AAV9 et contrôlée par un promoteur spécifique des cardiomyocytes. Dans des coupes précises de cœurs humains en insuffisance, ce vecteur a réussi à augmenter les niveaux de KLF15 et à améliorer la performance contractile sur plusieurs jours en culture.
Une feuille de route pour une thérapie génique plus douce
Pour un non-spécialiste, le message central est que ce travail montre comment augmenter avec précision un seul régulateur protecteur à l’intérieur des cellules musculaires cardiaques peut à la fois stabiliser leur identité et envoyer des signaux qui limitent la cicatrisation. En utilisant un activateur CRISPR qui ne coupe pas l’ADN, l’approche module finement les gènes endogènes du cœur plutôt que d’insérer un gène artificiel. L’étude définit une voie TGF-β → KLF15 → AZGP1 reliant le stress mécanique au remodelage délétère et démontre, chez la souris, dans des modèles cellulaires humains et dans des tranches de tissu cardiaque humain, que la restauration de KLF15 peut interrompre cette réaction en chaîne. Bien qu’encore au stade préclinique, le système CRISPRa compact ciblant les cardiomyocytes présenté ici offre une feuille de route potentielle pour traiter les formes courantes et non génétiques d’insuffisance cardiaque en reprogrammant l’activité génique plutôt qu’en réécrivant le génome.
Citation: Schoger, E., Kim, R., Bleckwedel, F. et al. Enhancing KLF15 activity in cardiomyocytes: a novel approach to prevent pathological reprogramming and fibrosis via nuclease-deficient dCas9VPR. Sig Transduct Target Ther 11, 76 (2026). https://doi.org/10.1038/s41392-026-02593-9
Mots-clés: insuffisance cardiaque, KLF15, activation CRISPR, fibrose cardiaque, AZGP1