Clear Sky Science · es
Generación molecular 3D con restricciones de interacción mediante un modelo de difusión posibilita la elaboración de farmacóforos basados en la estructura para el diseño de fármacos
Por qué es tan difícil diseñar mejores medicamentos
El descubrimiento moderno de fármacos a menudo depende de convencer a una pequeña molécula para que encaje en una proteína como una llave en una cerradura. Pero la llave debe hacer algo más que encajar: tiene que formar el conjunto correcto de pequeñas atracciones —como débiles tirones eléctricos y zonas que evitan el agua— para que el fármaco permanezca unido con fuerza y selectividad. El universo químico es astronómicamente grande, muy por encima de lo que contienen las bases de datos actuales, por lo que los investigadores buscan maneras más inteligentes de inventar nuevas llaves desde cero mientras preservan estos patrones de contacto cruciales.
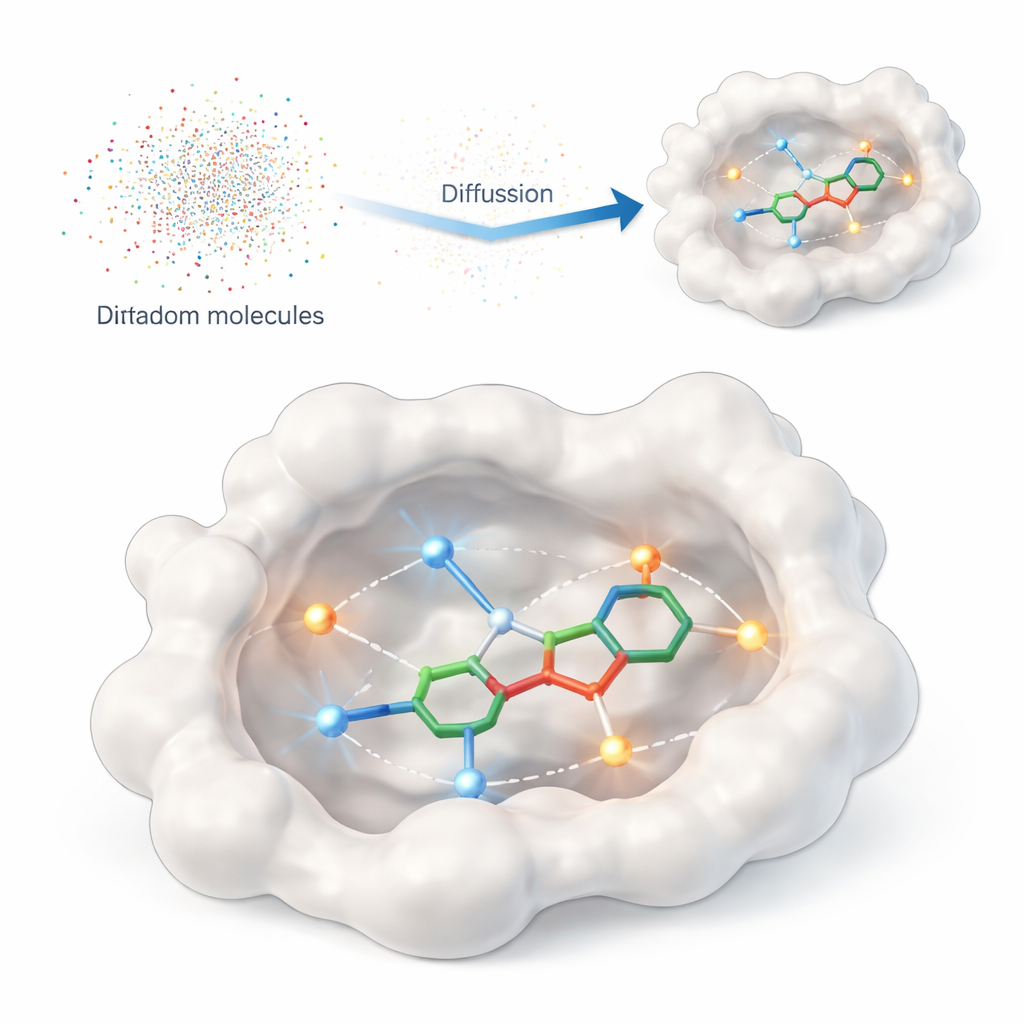
Enseñar a un ordenador qué es lo realmente importante
Este estudio presenta DiffPharma, un marco computacional que genera moléculas 3D con propiedades farmacológicas directamente dentro del sitio de unión de una proteína. En lugar de pedir al algoritmo que rastree enormes catálogos de compuestos existentes, DiffPharma crea nuevos compuestos átomo por átomo, guiado por cómo deben interactuar con la proteína. El método se basa en una clase moderna de modelos generativos llamados modelos de difusión, que parten de ruido aleatorio y lo “desruidan” gradualmente hasta lograr un objeto estructurado —en este caso, una molécula 3D alojada en el bolsillo proteico.
Codificar el apretón de manos de la proteína
Para indicar al modelo qué importa en la superficie de la proteína, los autores representan los contactos clave como pequeñas “partículas de interacción” esparcidas a lo largo de los trayectos entre la proteína y una molécula de referencia. Se enfatizan dos tipos comunes de interacción: los enlaces de hidrógeno, que actúan como imanes direccionales entre átomos específicos, y los contactos hidrófobos, donde las regiones oleosas se agrupan alejándose del agua. Redes neuronales separadas aprenden la geometría y la química de cada tipo de interacción, así como la forma general del bolsillo de unión, y luego una arquitectura de fusión especial combina estos puntos de vista en una única imagen coherente que guía la generación molecular.
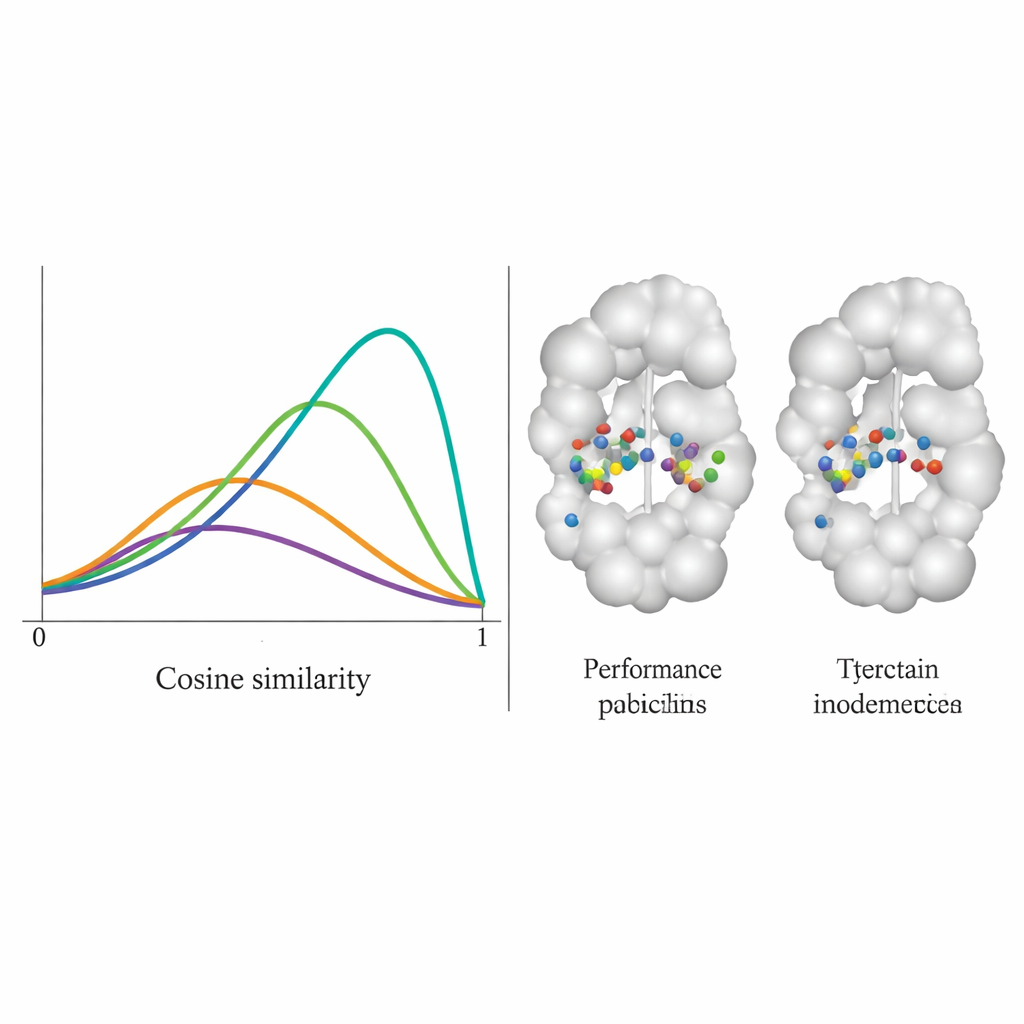
¿Hasta qué punto imita los patrones reales de unión?
El equipo probó DiffPharma en 100 pares distintos de proteína–molécula y evaluó cuán fielmente las nuevas moléculas reproducían los patrones de contacto originales, residuo por residuo. Midieron esto usando una puntuación de similitud por coseno entre 0 y 1, donde 1 significa concordancia perfecta. La distribución de DiffPharma se centró alrededor de 0,9, lo que significa que, en promedio, los mismos residuos proteicos formaron los mismos tipos de interacciones clave que en las estructuras de referencia —sustancialmente mejor que seis métodos competidores. De forma importante, el modelo hizo esto al tiempo que producía variedad en las formas moleculares, y los compuestos generados mantuvieron longitudes y ángulos de enlace realistas, así como la geometría 3D global típica de moléculas reales y estables.
De la teoría a candidatos prácticos de fármacos
Más allá de los puntos de referencia, los autores preguntaron si DiffPharma podía diseñar candidatos farmacológicos plausibles para objetivos reales. Para dos enzimas bien estudiadas —la quinasa AKT y una β‑lactamasa vinculada a la resistencia a antibióticos— el método generó moléculas que preservaron los patrones de interacción esenciales de ligandos conocidos y, a menudo, utilizaron andamiajes químicos distintos, una forma deseable de “salto de armazón” en química médica. En un estudio de caso más exigente sobre la proteasa principal del SARS‑CoV‑2, DiffPharma fue guiado mediante elecciones de interacción específicas y luego examinado con simulaciones de dinámica molecular y estimaciones de energía de unión. Las moléculas generadas bajo restricciones de interacción más estrictas formaron complejos más estables y en ocasiones mostraron energías de unión predichas más favorables que un inhibidor de referencia conocido. Notablemente, el sistema incluso redescubrió ese compuesto de referencia —a pesar de que nunca apareció en el entrenamiento— únicamente a partir de la estructura de la proteína y las instrucciones de interacción.
Qué significa esto para los medicamentos del futuro
Para un no especialista, DiffPharma puede entenderse como una herramienta de diseño inteligente y consciente del 3D para moléculas farmacológicas: dada la forma de un bolsillo proteico y un patrón deseado de “apretones de manos”, propone llaves químicamente razonables que encajan e interactúan de la manera correcta. Aunque aún no optimiza todas las propiedades que necesita un medicamento, como solubilidad o metabolismo, el método preserva de forma fiable el mapa de contactos crucial en la superficie proteica y explora nuevas regiones del espacio químico más allá de los catálogos actuales. Este enfoque guiado por interacciones puede ayudar a los investigadores a avanzar más rápidamente desde datos estructurales de proteínas relacionadas con enfermedades hacia puntos de partida diversos y realistas para el desarrollo experimental de fármacos.
Cita: Sako, M., Yasuo, N. & Sekijima, M. Interaction-constrained 3D molecular generation using a diffusion model enables structure-based pharmacophore modeling for drug design. npj Drug Discov. 3, 8 (2026). https://doi.org/10.1038/s44386-026-00040-x
Palabras clave: diseño de fármacos basado en la estructura, modelos generativos moleculares, modelado de farmacóforos, interacciones proteína–ligando, proteasa principal de SARS-CoV-2