Clear Sky Science · es
Acoplamiento competitivo guiado por IA para cribado virtual y predicción de la eficacia de compuestos
Búsquedas más inteligentes para nuevos medicamentos
Encontrar nuevos fármacos es un poco como buscar una aguja en un pajar formado por millones de moléculas. Este estudio muestra cómo los avances recientes en inteligencia artificial pueden hacer esa búsqueda más rápida y económica ayudando a los científicos a predecir qué moléculas tienen más probabilidades de adherirse a una proteína relacionada con la enfermedad y funcionar realmente como medicamentos. En lugar de probar un químico a la vez en el laboratorio, los autores utilizan modelos de IA para ejecutar concursos virtuales entre moléculas y dejar que los ganadores asciendan a la cima.
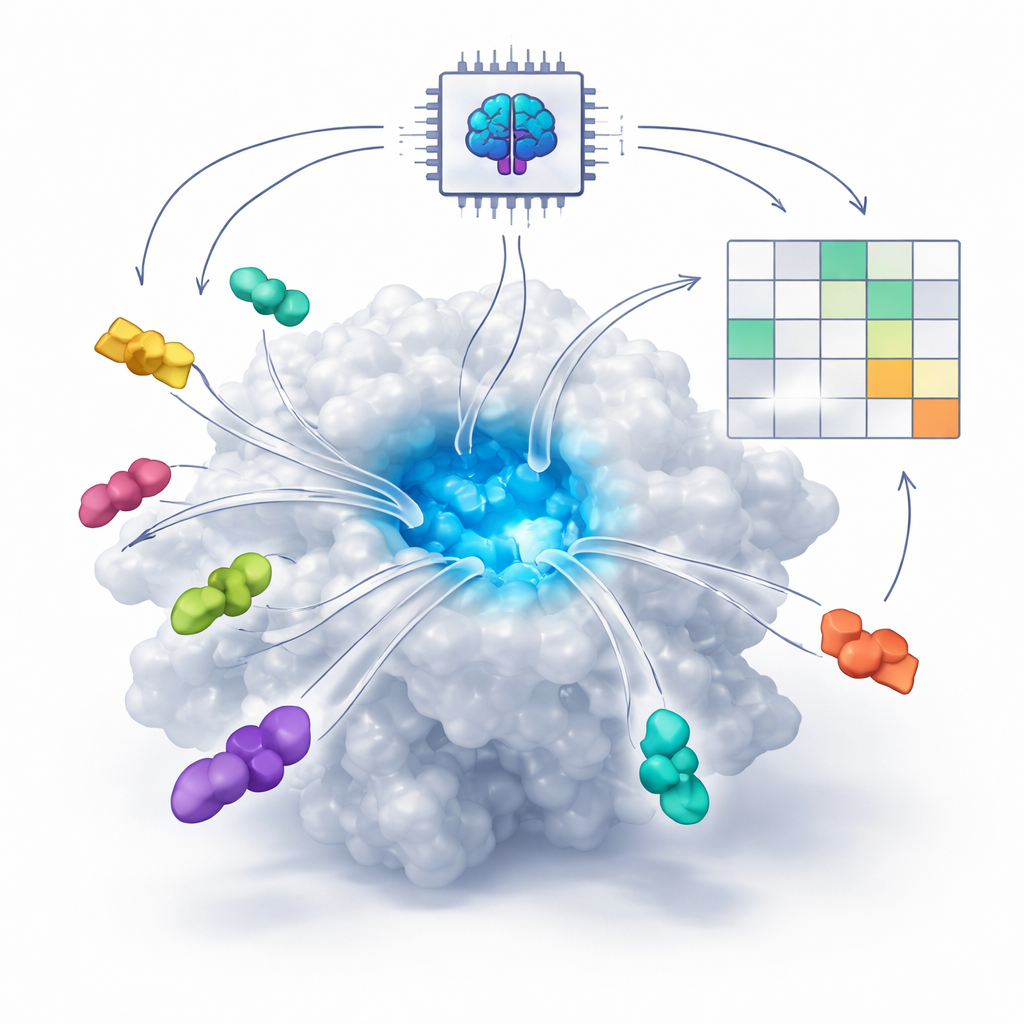
Cómo la IA aprende a ver los encajes de llave y cerradura molecular
Muchos fármacos modernos funcionan encajando en pequeños huecos de las proteínas, tal como una llave encaja en una cerradura. Tradicionalmente, los programas informáticos intentaban predecir este encaje usando ecuaciones físicas que estiman las fuerzas entre átomos. En los últimos años, sin embargo, nuevos sistemas de IA basados en difusión y co-plegamiento —como AlphaFold3 y Boltz— han aprendido a partir de un gran número de estructuras proteína–molécula conocidas. Estos sistemas ahora pueden “imaginar” cómo una proteína y un posible fármaco podrían plegarse juntos en tres dimensiones, incluso cuando no existe una estructura experimental. La pregunta central que abordan los autores es si estas herramientas de IA pueden hacer algo más que dibujar imágenes plausibles: ¿pueden también distinguir buenos fármacos de los malos?
Uniones reales frente a impostoras
El equipo probó primero 16 proteínas bien estudiadas más una enzima bacteriana más compleja llamada girasa del ADN. Para cada proteína, pidieron a los modelos de IA que colocaran tanto inhibidores activos conocidos como un conjunto de moléculas “fuera de objetivo” no relacionadas en el mismo sitio de unión. En lugar de confiar en una sola predicción, examinaron cuán consistentemente la IA colocaba cada molécula a lo largo de muchas ejecuciones. Los inhibidores reales tendían a volver al mismo lugar y orientación una y otra vez, agrupándose dentro de unas pocas billonésimas de metro entre sí. Las moléculas inactivas vagaban más y con frecuencia se situaban más lejos del bolsillo. Esta idea simple —convergencia de la pose— resultó ser una señal potente de que un compuesto encaja realmente con su objetivo proteico.
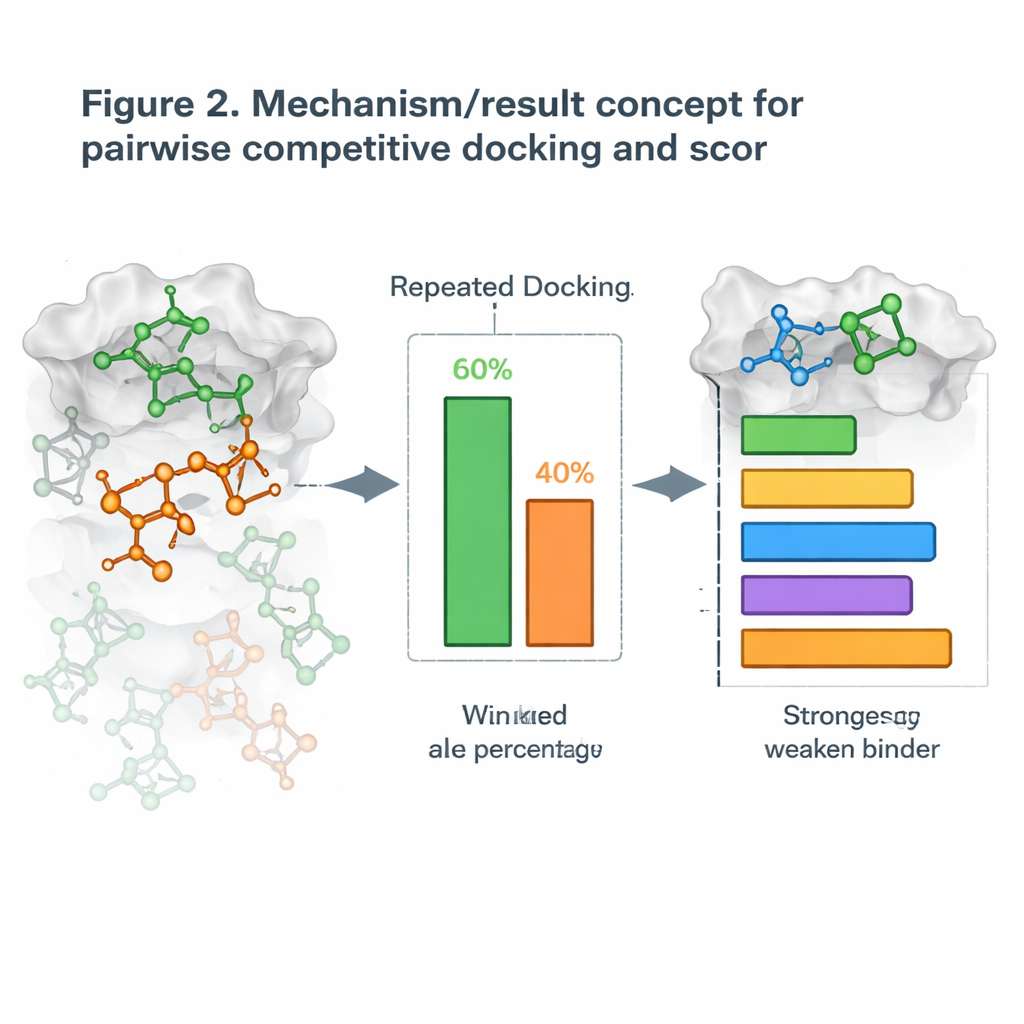
Convertir el acoplamiento en competición cara a cara
Partiendo de esto, los autores introdujeron una nueva estrategia que llaman acoplamiento competitivo por pares. En lugar de acoplar una molécula a la vez, acoplan dos candidatos simultáneamente con la proteína y los dejan “competir” por el mismo hueco. Tras muchas ejecuciones repetidas, se declara ganador del enfrentamiento la molécula que ocupa el sitio con más frecuencia. Al ejecutar todos los emparejamientos posibles, construyen una tabla de victorias y derrotas y calculan una Puntuación de Acoplamiento Competitivo para cada molécula, similar a clasificar jugadores en un torneo todos contra todos. Cuando estas puntuaciones se compararon con mediciones del mundo real sobre la fuerza con la que las moléculas bloquean sus objetivos, las clasificaciones a menudo coincidieron bien, con algunos sistemas proteicos mostrando una concordancia casi perfecta.
Del cribado virtual al diseño de mejores antibióticos
La girasa del ADN, una enzima esencial para las bacterias, sirvió como caso de prueba detallado. Esta proteína tiene varios bolsillos dirigidos por distintas clases de antibióticos, incluidas las fluoroquinolonas de uso generalizado. Los modelos de IA normalmente pudieron colocar cada clase de fármacos en su bolsillo correcto, y las puntuaciones de acoplamiento competitivo siguieron en líneas generales sus potencias medidas. Los autores ampliaron luego el enfoque a un cribado virtual de más de 3.000 fármacos aprobados, preguntando qué moléculas competían mejor por el sitio de las fluoroquinolonas. Su estrategia en dos pasos —primero usar la competencia “todo a la vez” para seleccionar probables ganadores, y luego filtrar según cuán estrechamente se agrupaban en el bolsillo— enriqueció considerablemente las verdaderas fluoroquinolonas mientras descartaba candidatos más débiles. Finalmente, usaron un generador de moléculas impulsado por IA para proponer nuevas estructuras tipo fluoroquinolona y aplicaron el acoplamiento competitivo para encontrar un puñado con predicciones de unión incluso mejores y propiedades semejantes a fármacos aceptables.
Promesas, límites y lo que significa para los pacientes
El estudio demuestra que los modelos modernos de IA pueden hacer más que dibujar estructuras proteína–fármaco plausibles: cuando se ejecutan en un marco competitivo, pueden ayudar a ordenar compuestos de una manera que a menudo refleja los datos experimentales reales. Esto no reemplaza el trabajo de laboratorio: el rendimiento sigue dependiendo en gran medida de la proteína particular, algunos bolsillos se predicen mal y los modelos de IA pueden fallar con moléculas muy grandes o inusuales. Pero a medida que estos modelos y sus datos de entrenamiento mejoren, enfoques como el acoplamiento competitivo por pares podrían hacer que el descubrimiento temprano de fármacos sea mucho más eficiente. Para los pacientes, eso podría traducirse eventualmente en un desarrollo más rápido de medicamentos dirigidos, incluidos nuevos antibióticos que mantengan el ritmo frente a bacterias resistentes.
Cita: Mirgaux, M., Barcelli, V., Chua, A.C.Y. et al. AI-guided competitive docking for virtual screening and compound efficacy prediction. npj Drug Discov. 3, 6 (2026). https://doi.org/10.1038/s44386-026-00039-4
Palabras clave: descubrimiento de fármacos con IA, cribado virtual, acoplamiento molecular, unión proteína-ligando, diseño de antibióticos