Clear Sky Science · es
Computación científica eficiente en energía usando reservorios químicos
Por qué convertir la química en computación importa
Los superordenadores modernos consumen cantidades enormes de electricidad para simular el clima, diseñar nuevos fármacos o entrenar inteligencia artificial. A medida que nos acercamos a los límites físicos de los chips tradicionales, obtener más rendimiento por vatio es cada vez más difícil y caro. Este artículo explora un camino radicalmente distinto: usar reacciones químicas reales como el motor de la computación científica. Tratando las moléculas y sus interacciones como las piezas móviles de un ordenador, los autores describen cómo máquinas futuras podrían resolver ecuaciones complejas con mucha menos energía que el hardware digital actual.
De las células vivas a las calculadoras químicas
Las células vivas son expertas en resolver problemas. Constantemente gestionan miles de reacciones para adaptarse, crecer y sobrevivir, todo ello empleando sorprendentemente poca energía. En el centro de este comportamiento están las redes de reacciones químicas: reacciones interconectadas cuyas tasas y concentraciones varían en el tiempo. Estas redes pueden describirse mediante ecuaciones diferenciales ordinarias, el mismo lenguaje matemático usado para modelar desde epidemias hasta flujos turbulentos. La idea central de este trabajo es que si la química ya sigue estas ecuaciones, podríamos aprovecharla directamente para realizar los cálculos que hoy ejecutan los chips de silicio.
Cómo las ecuaciones se convierten en redes de reacciones
Los autores presentan ChemComp, un marco de software que toma un sistema de ecuaciones diferenciales y lo convierte sistemáticamente en una red abstracta de reacciones. ChemComp utiliza tecnología moderna de compiladores para descomponer un problema matemático en patrones que pueden representarse mediante reacciones idealizadas, y luego las organiza en una red con especies, conexiones y constantes de reacción bien definidas. Estas reacciones abstractas aún no corresponden a moléculas reales, pero constituyen un plano para un ordenador químico. El marco puede después buscar en bases de datos bioquímicas motivos de reacción reales que se comporten de forma similar, priorizando opciones prácticas, seguras y potencialmente eficientes en energía en un entorno de laboratorio.
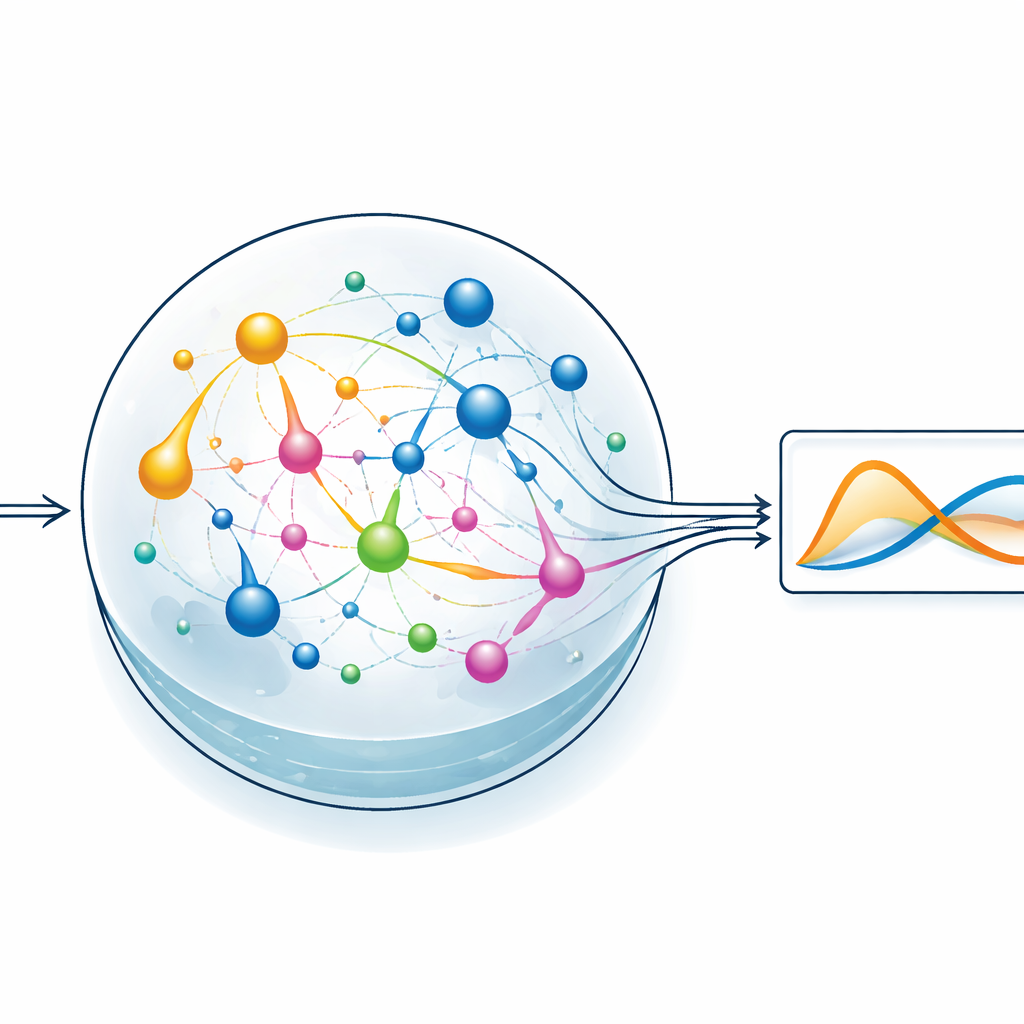
Dejar que un reservorio químico haga el trabajo duro
Para poner a prueba la idea, el equipo se centra en un estilo de aprendizaje automático llamado computación por reservorio. Aquí, un sistema dinámico fijo transforma una señal de entrada en un patrón interno rico y enmarañado, y sólo se entrena una capa de lectura simple para producir la salida deseada. En la versión de ChemComp, el reservorio es un conjunto de reacciones en un vaso bien agitado; las concentraciones cambiantes de las sustancias químicas constituyen los estados internos. Los autores compilan un sistema clásico de dos variables conocido como el modelo de Sel’kov–Schnakenberg —originalmente usado para estudiar oscilaciones en el metabolismo— en redes de reacciones candidatas. Luego simulan cómo responden estas redes con el tiempo cuando son impulsadas por flujos de entrada y salida de sustancias en el vaso, y usan regresión lineal básica para combinar las trazas de concentración en una aproximación de la solución objetivo.
Probando redes químicas sencillas y más ricas
Los investigadores comparan dos reservorios candidatos: uno con sólo dos especies químicas y dos reacciones, y otro con cinco especies y cinco reacciones. A ambas redes se les asignan concentraciones iniciales y tasas de flujo adecuadas, y luego se simulan en funcionamiento. Incluso el sistema más pequeño puede reproducir de forma aproximada el comportamiento oscilatorio de las ecuaciones objetivo, pero la red mayor lo hace notablemente mejor, reduciendo el error tanto en entrenamiento como en prueba. Al explorar diferentes concentraciones iniciales y constantes de velocidad de reacción, los autores trazan regiones donde el sistema químico se ajusta más estrechamente a la dinámica deseada. Cada reacción actúa, en efecto, como una función base en un problema de ajuste de curvas: cuantas más reacciones variadas estén disponibles, más fácil es aproximar comportamientos complejos, a costa de una mayor complejidad del sistema.
Camino hacia la computación de bajo consumo en el laboratorio
Más allá de las simulaciones, el artículo avanza hacia dispositivos prácticos. Discute cómo la elección de reacciones debe equilibrar el uso de energía, la capacidad de control mediante enzimas o catalizadores y la posibilidad de medir especies clave en tiempo real, por ejemplo con métodos ópticos o electroquímicos. Los autores sugieren que futuras plataformas microfluídicas podrían albergar redes de reacciones cuidadosamente seleccionadas, con control espacial de entradas y sensores integrados. Aunque quedan muchos desafíos de ingeniería —desde mapear ecuaciones a química real hasta manejar el ruido y los límites de medición—, el estudio muestra que sistemas de reacción modestos ya pueden emular soluciones de ecuaciones diferenciales acopladas. Para el lector general, el mensaje principal es que la química misma puede actuar como un ordenador analógico, abriendo un camino hacia cálculos científicos que aprovechan los procesos energéticamente eficientes que la naturaleza ha perfeccionado durante miles de millones de años.
Cita: Johnson, C.G.M., Bohm Agostini, N., Cannon, W.R. et al. Energy-efficient scientific computing using chemical reservoirs. npj Unconv. Comput. 3, 17 (2026). https://doi.org/10.1038/s44335-026-00053-9
Palabras clave: computación química, computación eficiente en energía, computación por reservorio, redes de reacciones químicas, ecuaciones diferenciales ordinarias