Clear Sky Science · es
Un marco de extremo a extremo para la reactividad en catálisis heterogénea
Por qué importa diseñar catalizadores más rápido
La sociedad moderna depende de los catalizadores para fabricar combustibles, plásticos, fertilizantes y multitud de productos cotidianos. Sin embargo, encontrar catalizadores mejores a menudo equivale a buscar una aguja en un pajar, porque cada material puede promover miles de reacciones microscópicas posibles a la vez. Este artículo presenta CARE, un nuevo marco computacional que emplea reglas inteligentes y aprendizaje automático para cartografiar y simular estas enmarañadas redes de reacción mucho más rápido y de forma más completa que antes. Al hacerlo, promete orientar tecnologías energéticas más limpias y procesos químicos más eficientes mientras reduce drásticamente los costes computacionales.
Desenredando vías de reacción abarrotadas
En la superficie de un catalizador sólido, las moléculas entrantes no siguen simplemente una única ruta clara de reactivo a producto. En su lugar, transitan por un laberinto de intermedios fugaces y vías en competencia. Los métodos computacionales tradicionales dependen de la intuición humana para seleccionar un conjunto limitado de pasos posibles y luego usan cálculos cuánticos para evaluar sus energías. Esto funciona para redes pequeñas pero se desmorona rápidamente a medida que los sistemas se vuelven más complejos, pasando por alto rutas raras que pueden gobernar la actividad a largo plazo, la desactivación o la selectividad. CARE aborda este desafío construyendo automáticamente redes de reacción muy extensas a partir de reglas de construcción simples, garantizando que se incluyan todos los eventos plausibles de ruptura y formación de enlaces entre carbono, hidrógeno y oxígeno, incluso aquellos que normalmente los químicos podrían descartar.
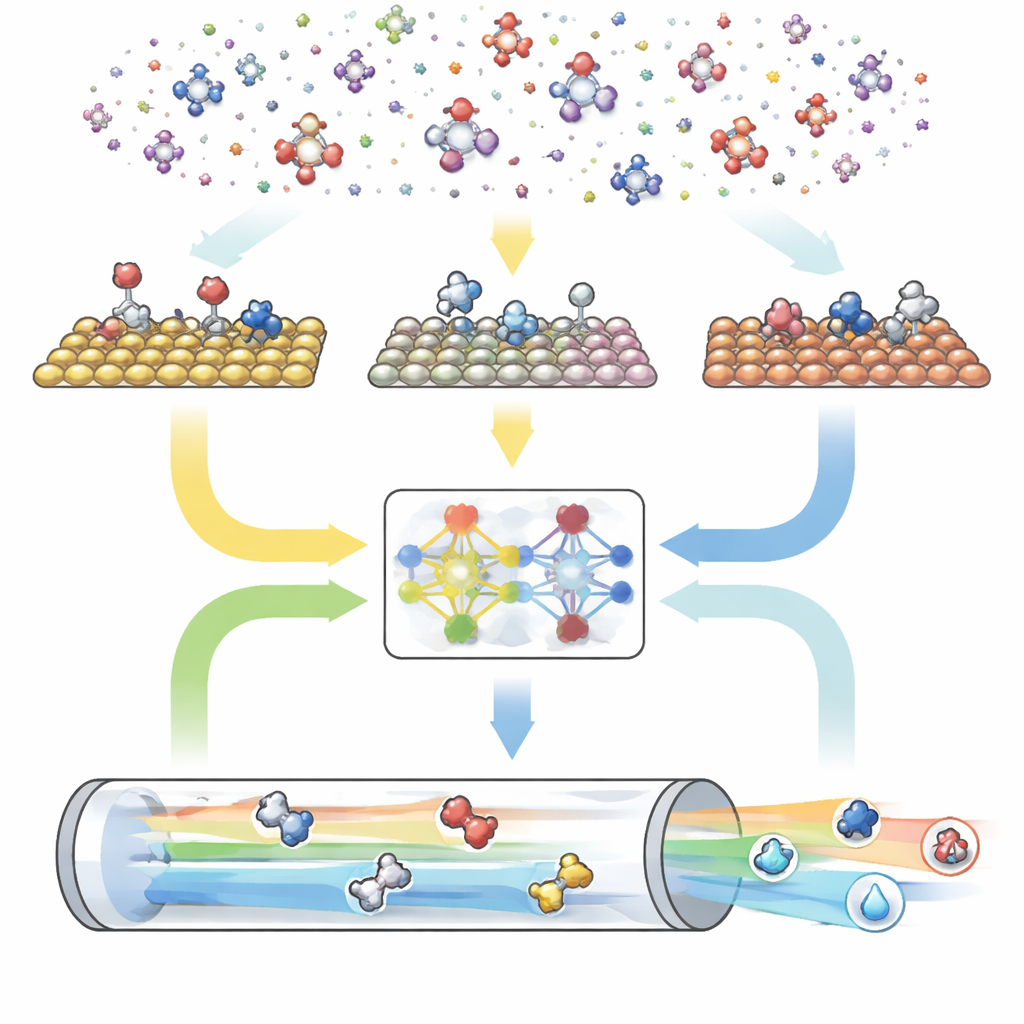
Un motor digital de tres partes para reacciones
CARE está diseñado como una canalización integral con tres módulos principales. Primero, un generador basado en reglas define el “espacio químico” eligiendo el número máximo de átomos de carbono y oxígeno y aplicando luego plantillas simples para crear todas las moléculas coincidentes y sus formas enlazadas a la superficie. Segundo, un módulo de evaluación energética invoca modelos modernos de aprendizaje automático—especialmente una red neuronal de grafos llamada GAME-Net-UQ—para estimar las energías de intermedios y estados de transición en numerosas superficies metálicas. Este modelo trata cada estructura como una red de átomos y enlaces, devuelve tanto una energía como una incertidumbre, y es preciso hasta unas pocas décimas de electrónvoltio mientras sigue siendo ligero y rápido. Tercero, un solucionador microcinético utiliza estas energías para calcular cómo progresan todas las reacciones conjuntamente bajo condiciones realistas de temperatura, presión, potencial y pH, prediciendo tasas de reacción globales, coberturas superficiales y selectividad de productos.
Pruebas reales: moléculas de combustible y química climática
Para demostrar que CARE no es solo un ejercicio teórico, los autores lo aplican a tres problemas industrialmente relevantes de dificultad creciente. Para la descomposición del metanol—una reacción importante para el almacenamiento de hidrógeno—generan una red modesta y la evalúan en muchos catalizadores metálicos y caras cristalinas. CARE reproduce la familiar tendencia en “volcán” de la actividad e identifica correctamente al rutenio como uno de los mejores desempeños, en concordancia con experimentos, pero gastando una fracción mínima del tiempo de cálculo requerido por cálculos cuánticos completos. A continuación, pasan a la conversión electroquímica de dióxido de carbono en cobre, centrándose en cómo surgen productos de tres carbonos como el 1-propanol y el propileno. Al incluir pasos especiales que tienen en cuenta protones, electrones y condiciones de solución, CARE captura cómo el pH y el voltaje aplicado desplazan las vías y predice correctamente que el 1-propanol se favorece sobre el propileno, reflejando estudios detallados previos.
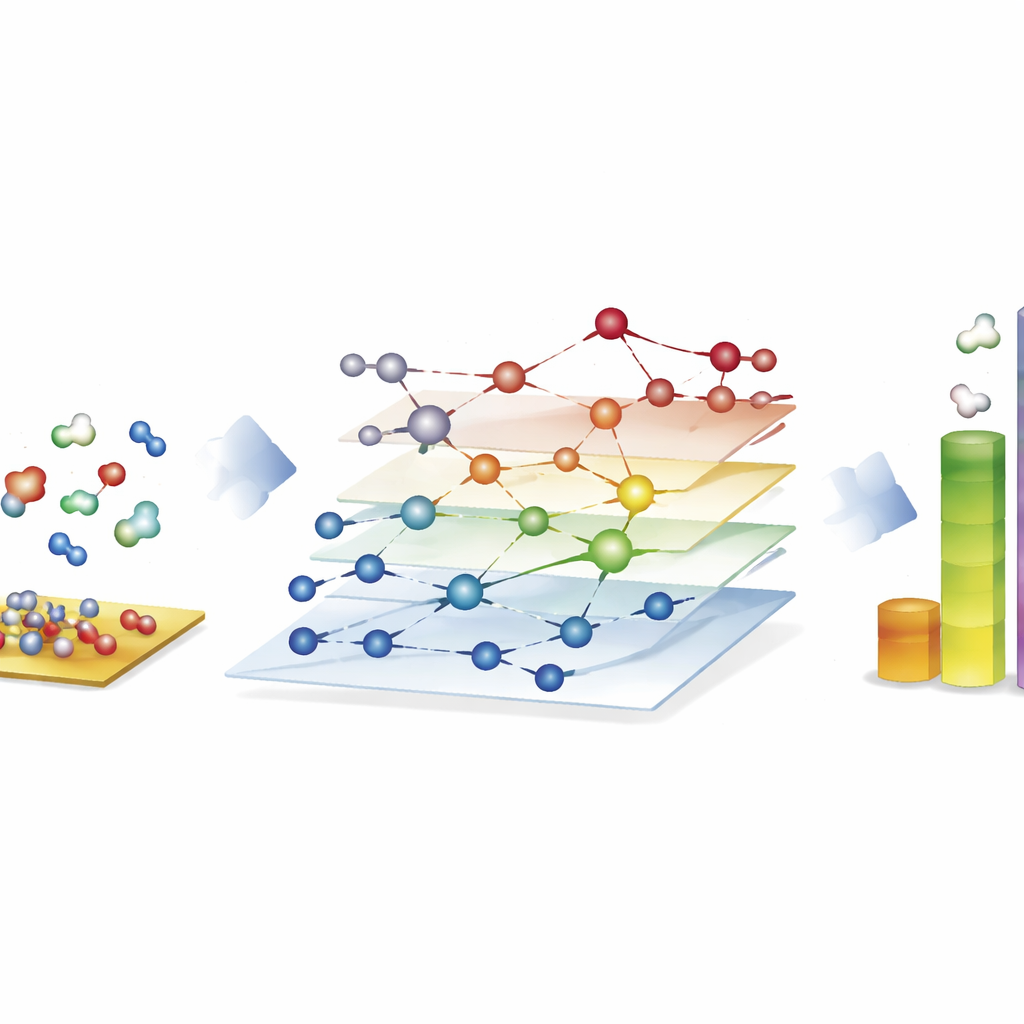
Explorando enormes redes de reacción para combustibles sintéticos
La demostración más llamativa proviene del proceso de Fischer–Tropsch, que convierte mezclas de monóxido de carbono e hidrógeno en hidrocarburos de cadena larga para combustibles y productos químicos. Aquí, los autores construyen redes con casi 40.000 especies superficiales y alrededor de 370.000 reacciones elementales—muy por encima de lo que los estudios tradicionales basados en métodos cuánticos pueden explorar completamente. Usando CARE, evalúan todos los intermedios y las barreras reactivas clave en superficies de cobalto, hierro, níquel y rutenio en solo unas horas en hardware estándar, una aceleración de aproximadamente un millón de veces respecto a cálculos cuánticos directos. Las simulaciones microcinéticas en estas redes reproducen tendencias conocidas: cobalto e hierro forman preferentemente cadenas de hidrocarburos más largas, el hierro produce más dióxido de carbono mediante reacciones secundarias, y el níquel tiende a una hidrogenación más intensa. Aunque algunos detalles, como los rendimientos de metano, siguen siendo imperfectos, el marco revela qué pasos de formación de enlaces dominan el crecimiento de cadenas y destaca dónde los modelos aún necesitan perfeccionarse.
Qué significa esto para los catalizadores del futuro
Para los no especialistas, el mensaje clave es que CARE proporciona una forma práctica de explorar espacios de reacción enormes en superficies catalíticas que antes estaban fuera de alcance. Al automatizar la generación de redes, conectar modelos “sustitutos” rápidos de aprendizaje automático para energías cuánticas y resolver la cinética resultante de forma eficiente, puede clasificar catalizadores candidatos, identificar condiciones de operación prometedoras y descubrir vías inesperadas con mucho menos sesgo humano y gasto computacional. Aunque los autores señalan desafíos restantes—como un mejor tratamiento de superficies abarrotadas, efectos de solvente e incluso redes de mayor tamaño—el trabajo apunta a un futuro en el que los ordenadores puedan cribar rápidamente reacciones complejas, desde la reducción de dióxido de carbono hasta el reciclado de plásticos y la valorización de la biomasa, guiando los experimentos hacia las ideas más prometedoras en lugar de dejar el descubrimiento al ensayo y error.
Cita: Morandi, S., Loveday, O., Renningholtz, T. et al. An end-to-end framework for reactivity in heterogeneous catalysis. Nat Chem Eng 3, 169–180 (2026). https://doi.org/10.1038/s44286-026-00361-8
Palabras clave: catálisis heterogénea, redes de reacción, aprendizaje automático, modelado microcinético, síntesis de Fischer–Tropsch