Clear Sky Science · es
Difusión condicional con alineación modal consciente de la localidad para generar conjuntos conformacionales proteicos diversos
Por qué el movimiento de las proteínas importa
Las proteínas en nuestras células no son esculturas rígidas; se comportan más bien como pequeñas máquinas flexibles que cambian constantemente de forma. Estos cambios de forma pueden controlar cómo las enzimas catalizan reacciones, cómo los receptores responden a fármacos y cómo fluyen las señales en las células. Sin embargo, la mayoría de las imágenes familiares de proteínas muestran solo una “instantánea” estructural, perdiendo el rico conjunto de formas que realmente existen. Este artículo presenta Mac-Diff, un método de inteligencia artificial que puede generar rápidamente muchas formas realistas para una proteína dada, ayudando a los científicos a ver no solo cómo es una proteína, sino cómo respira y se mueve.
De instantáneas únicas a conjuntos en movimiento
Durante décadas, los investigadores han confiado en experimentos minuciosos o en largas simulaciones de dinámica molecular para explorar el movimiento de las proteínas, ambos procesos lentos y costosos. Herramientas revolucionarias como AlphaFold2 ahora predicen la estructura 3D más probable de una proteína directamente a partir de su secuencia de aminoácidos, pero por lo general devuelven solo una o pocas formas preferidas. Muchas proteínas, especialmente las implicadas en señalización y regulación alostérica, ocupan de forma natural múltiples estados vagamente definidos. Los autores sostienen que, para entender cómo funcionan realmente estas proteínas —y para diseñar fármacos que se unan a formas transitorias y menos evidentes— necesitamos una forma de generar conjuntos completos de conformaciones plausibles, no solo una mejor conjetura.
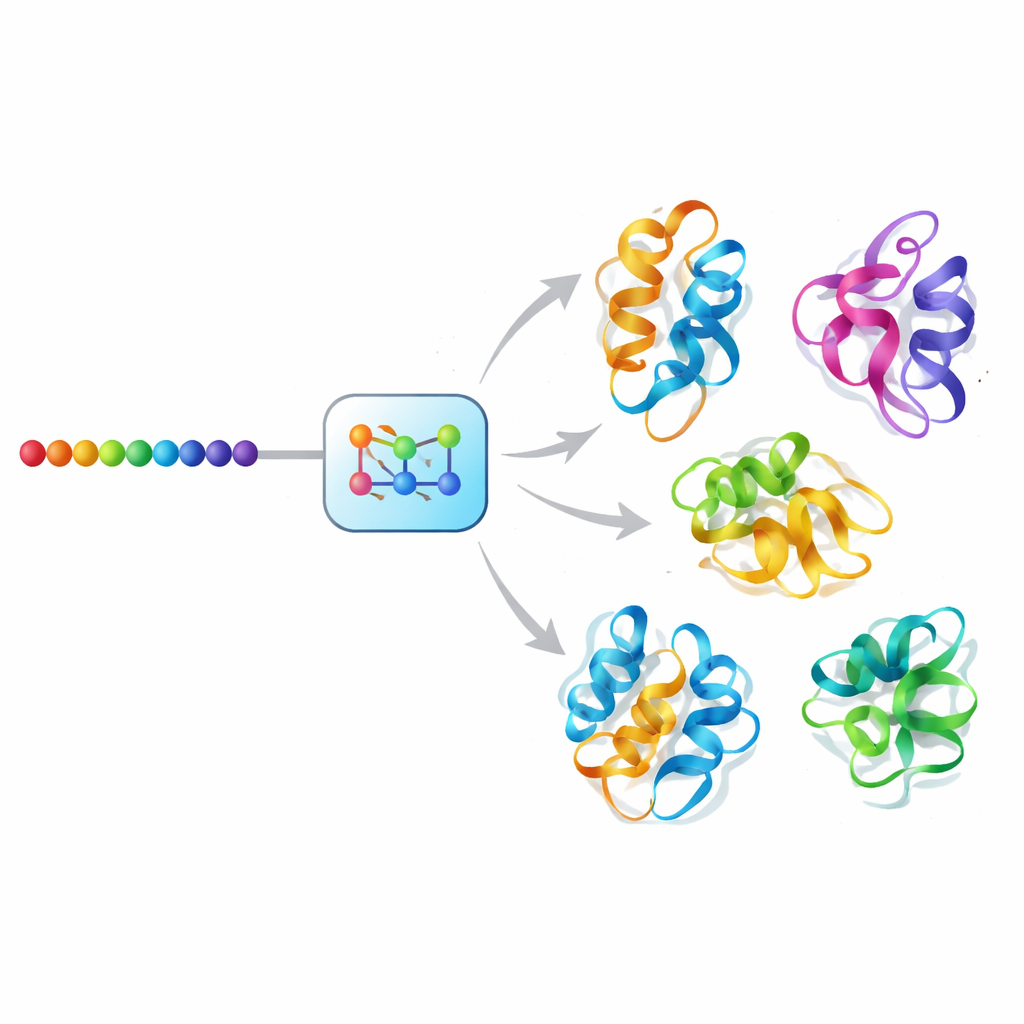
Un enfoque de "difusión" por IA para el movimiento proteico
Mac-Diff afronta este desafío usando un modelo generativo de estilo difusión, una clase de IA que ha impulsado avances recientes en síntesis de imágenes. En lugar de eliminar ruido de fotografías, Mac-Diff elimina ruido de descripciones geométricas abstractas de las espinas dorsales proteicas. El modelo representa una proteína como una cuadrícula de relaciones por pares entre sus residuos —distancias y ángulos que son insensibles a cómo se rota o traslada toda la molécula. En un paso directo, el sistema añade gradualmente ruido a estos patrones geométricos hasta que parecen estática aleatoria. En el paso inverso, aprende a eliminar el ruido paso a paso, guiado por la secuencia de aminoácidos de la proteína, hasta que reaparecen geometrías coherentes compatibles con 3D, que luego pueden convertirse en modelos atómicos completos mediante software estándar de construcción de estructuras.
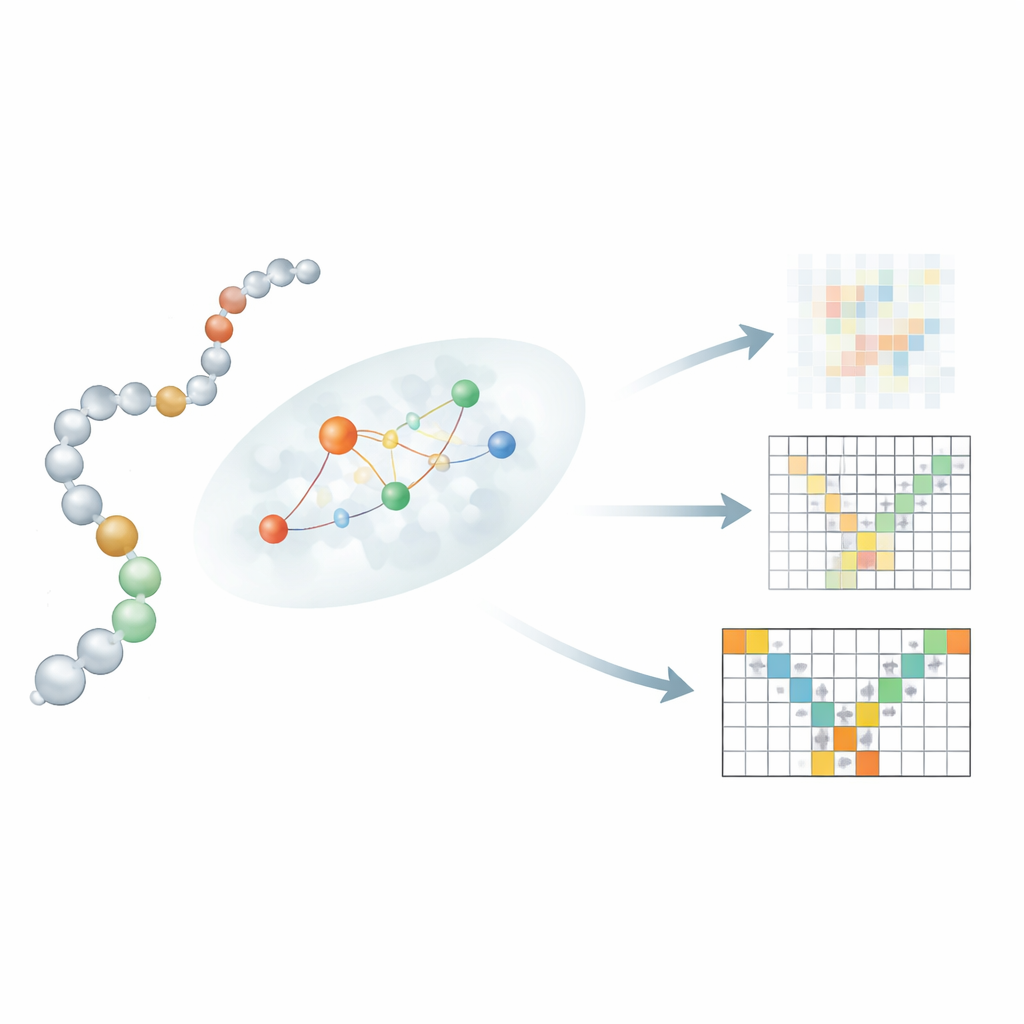
Permitir que la secuencia hable con la estructura localmente
Una innovación clave reside en cómo Mac-Diff conecta una secuencia lineal de residuos con sus vecinos tridimensionales. Permitir simplemente que cada residuo atienda a todos los demás residuos, como en modelos de texto a imagen, difuminaría restricciones físicas importantes. En su lugar, los autores introducen un mecanismo de atención "consciente de la localidad" que centra cada residuo en un pequeño vecindario probable de socios de interacción. Para estimar estos vecindarios, Mac-Diff usa tres ingredientes: un modelo de lenguaje proteico llamado ESM-2 que codifica el contexto bioquímico de cada residuo; un mapa de contactos que insinúa qué pares de residuos probablemente estén próximos; y una regla simple que favorece residuos cercanos a lo largo de la cadena. Estas señales se combinan para que, durante la desruidosificación, el modelo utilice preferentemente información de residuos que son socios físicamente plausibles, afinando su capacidad para reconstruir estructuras realistas y flexibles.
Evaluación frente a simulaciones largas y proteínas cambiantes de forma
Los investigadores evaluaron Mac-Diff en dos frentes exigentes. Primero, preguntaron si podía reproducir la amplia distribución de formas observada en simulaciones de dinámica molecular largas y cuidadosamente calculadas de proteínas de plegamiento rápido y de una proteína de referencia clásica conocida como BPTI. En varias medidas que comparan propiedades estadísticas de los conjuntos generados con los datos de simulación —como distribuciones de distancias dentro de la proteína y la compacidad global— Mac-Diff igualó o superó a métodos de IA competidores, al tiempo que generó una mayor variedad de conformaciones. Capturó la mayor parte de los estados “metaestables” clave identificados en las simulaciones y reprodujo los patrones de flexibilidad a nivel de residuo con alta correlación, lo que indica que sus conjuntos reflejan tanto los pliegues globales como los movimientos locales de forma realista.
Revelando estados funcionales ocultos
En segundo lugar, el equipo desafió a Mac-Diff con proteínas conocidas por adoptar formas muy diferentes mientras realizan su función, incluida la adenilato-cinasa, que alterna entre formas abiertas y cerradas durante el metabolismo energético, y un conjunto curado de 40 proteínas con dos conformaciones determinadas experimentalmente cada una. Mac-Diff generó solo 100 estructuras candidatas por proteína —mucho menos que las trayectorias típicas de simulación— y aun así recuperó la mayoría de los estados conocidos con buen acuerdo geométrico. En la adenilato-cinasa, por ejemplo, produjo conformaciones tanto abiertas como cerradas con alta similitud a estructuras cristalográficas, mientras que varios métodos populares tendían a favorecer solo un estado. El modelo también se ejecutó unas mil veces más rápido que las simulaciones convencionales en hardware comparable, lo que hace que la exploración sistemática de la diversidad de formas sea mucho más práctica.
Qué significa esto para la biología y la medicina
En términos cotidianos, Mac-Diff convierte la secuencia de una proteína en una galería de posturas plausibles en lugar de un único retrato, y lo hace con aprecio por qué partes es probable que se empujen o se ciñan entre sí en 3D. Al muestrear estos conjuntos con precisión y eficiencia, el método ofrece una forma de sondear cómo los cambios sutiles de forma sustentan la función, identificar conformaciones raras pero importantes y buscar bolsillos de unión a fármacos que aparecen solo en estados transitorios. Aunque aún no captura las películas ordenadas en el tiempo que proporcionan las simulaciones, Mac-Diff acerca el paisaje dinámico de las proteínas para muchos más sistemas, prometiendo nuevas ideas en biología estructural, diseño de fármacos e ingeniería de proteínas.
Cita: Wang, B., Wang, C., Chen, J. et al. Conditional diffusion with locality-aware modal alignment for generating diverse protein conformational ensembles. Nat Mach Intell 8, 415–434 (2026). https://doi.org/10.1038/s42256-026-01198-9
Palabras clave: dinámica de proteínas, modelos de difusión, conjuntos conformacionales, proteínas alostéricas, descubrimiento de fármacos