Clear Sky Science · es
Robustez sin precedentes de modelos de energía atómica informados por la física a y por encima de la temperatura ambiente
Por qué importa esto para la química cotidiana
Las simulaciones por ordenador son las herramientas de trabajo de la química y la ciencia de materiales modernas. Permiten a los científicos ver cómo las moléculas se retuercen, vibran y colisionan in silico en lugar de recurrir a experimentos costosos y que consumen mucho tiempo. Pero cuando estas simulaciones dependen del aprendizaje automático, pueden de pronto "explotar", produciendo formas moleculares imposibles, especialmente a temperaturas más altas. Este estudio presenta un nuevo tipo de modelo de aprendizaje automático informado por la física que puede ejecutar dichas simulaciones durante tiempos extremadamente largos, a temperaturas de hasta 1000 kelvin, sin venirse abajo.
De atajos ingeniosos a simulaciones frágiles
La química cuántica tradicional calcula las energías moleculares con gran exactitud pero es dolorosamente lenta. Los campos de fuerza más simples son rápidos pero a menudo aproximados. Los potenciales aprendidos por máquina pretenden combinar lo mejor de ambos mundos: aprenden un atajo desde la geometría molecular hasta la energía y las fuerzas, y luego usan ese atajo para impulsar la dinámica molecular. En papel, muchos de esos modelos parecen excelentes, presumiendo de errores medios diminutos en conjuntos de prueba estándar. En la práctica, esos números pueden ser engañosos. Cuando las moléculas exploran nuevas conformaciones durante una simulación—especialmente a temperaturas altas—muchos modelos se ven forzados fuera del rango de estructuras con las que se entrenaron. En lugar de dirigir suavemente a las moléculas de vuelta a formas realistas, pueden predecir fuerzas que estiran o aplastan enlaces hasta que todo el sistema resulta no físico y la simulación colapsa.
Construir modelos sobre bloques cuánticos
Los autores abordan esta fragilidad cambiando qué aprende el modelo y cómo se guía por la física previa. Usan un marco llamado FFLUX, que se basa en el enfoque de Átomos Cuánticos Interactuantes (IQA). En IQA, una molécula se divide en "átomos topológicos" cuyas energías individuales se determinan directamente a partir de la mecánica cuántica. Esas energías atómicas tienen significado físico y suman la energía total de la molécula. En lugar de aprender energías de sitio arbitrarias, los nuevos modelos de proceso Gaussiano aprenden estas energías atómicas de raíz cuántica, proporcionando un anclaje físico profundo para cada predicción. Cuatro moléculas orgánicas flexibles—glicina y serina con péptido cap, malondialdehído y aspirina—sirven como bancos de prueba desafiantes por sus numerosos movimientos internos y la conocida dificultad que plantean a los campos de fuerza aprendidos por máquina existentes.
Enseñar al modelo a esperar problemas
Una innovación clave reside en cómo se configura el proceso Gaussiano antes de que vea datos: su "función media", que codifica lo que el modelo asume en regiones poco conocidas. La mayoría de trabajos previos simplemente establecen esta media en cero, pretendiendo efectivamente que el modelo no tiene expectativas previas. Los autores, en cambio, desplazan deliberadamente esta media hacia estados de energía atómica más altos, manteniéndola físicamente sensata. Esta elección de diseño significa que cuando el modelo se ve obligado a extrapolar—por ejemplo, cuando los enlaces están temporalmente sobreestirados—prefiere de forma natural predicciones que penalizan distorsiones extremas. En pruebas extensas, versiones del modelo que solo diferían en este prior se comportaron de manera muy distinta. Los modelos con medias convencionales o de baja energía a menudo sobrevivían menos de un picosegundo antes de que una molécula explotara o implosionara. En contraste, la mejor media de alta energía (apodada MF5) produjo simulaciones que permanecieron estables durante toda la ventana de prueba de un nanosegundo a temperaturas de 300 a 1000 kelvin para las cuatro moléculas.
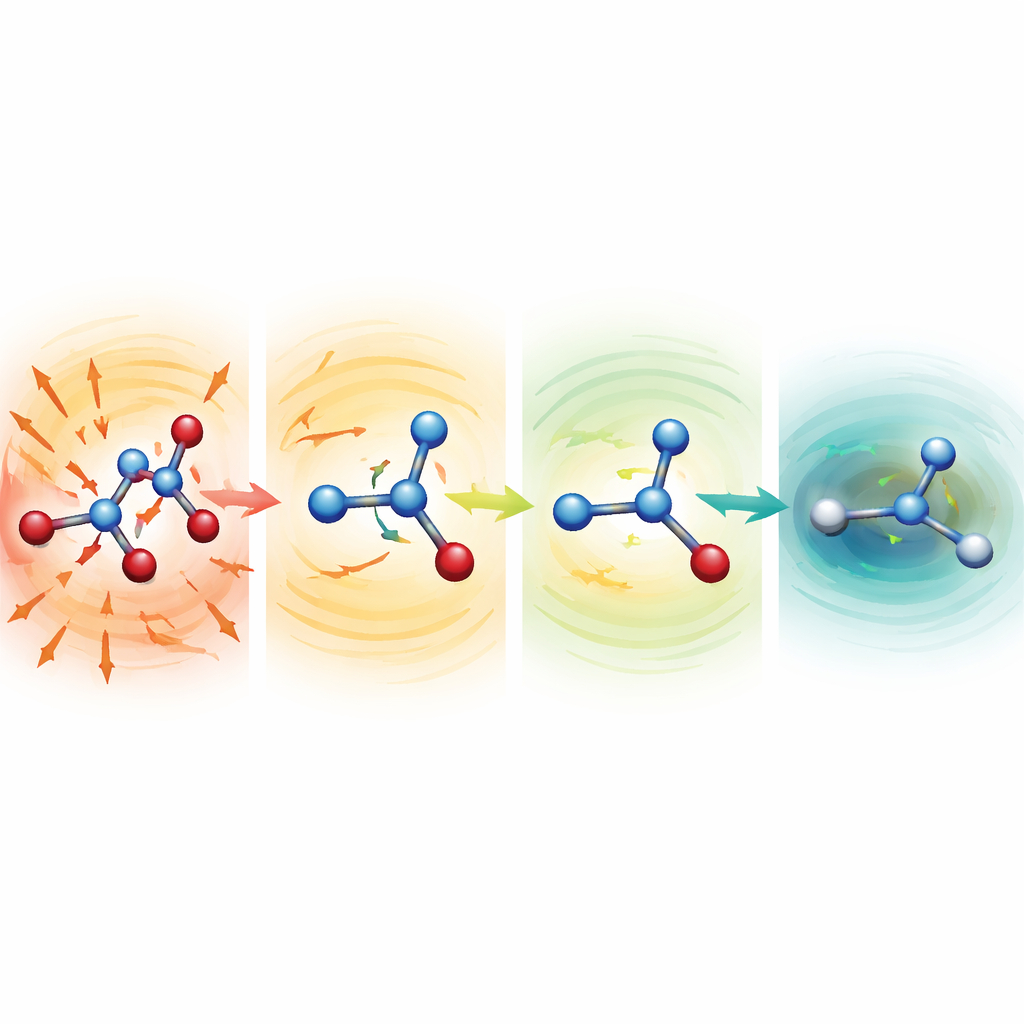
Ver cómo las moléculas distorsionadas se autocuran
Para indagar por qué funcionan tan bien los modelos robustos, los investigadores iniciaron simulaciones desde estructuras deliberadamente destrozadas con enlaces severamente estirados o comprimidos. Para serina, aspirina y malondialdehído, estos puntos de partida estaban cientos hasta más de mil kilocalorías por mol por encima de una estructura normal—configuraciones que normalmente serían desastrosas. Con funciones medias más débiles, las moléculas se desintegraron rápidamente. Con la configuración MF5, sin embargo, las fuerzas predichas señalaron inmediatamente en direcciones que acortaron los enlaces elongados y alargaron los comprimidos. En decenas a cientos de pasos de tiempo, las moléculas se relajaron hacia formas realistas y luego continuaron evolucionando de forma estable. El equipo también mostró que, sin haber sido entrenados jamás con fuerzas, esos mismos modelos pueden guiar optimizaciones de geometría de dipeptido de alanina, reproduciendo conformaciones de baja energía conocidas y energías relativas dentro de unas pocas décimas de kilocaloría por mol, pero a un coste aproximadamente 200 veces menor que los cálculos cuánticos completos.
Simulaciones largas y calientes en hardware corriente
La robustez no consiste solo en sobrevivir a un inicio difícil; consiste en aguantar durante millones o miles de millones de pasos de tiempo. Los autores llevaron sus mejores modelos más lejos ejecutando 50 simulaciones independientes a 500 kelvin, cada una de 10 nanosegundos, sobre las cuatro moléculas de prueba. Ninguna de estas ejecuciones colapsó, sumando un tiempo de simulación combinado de medio microsegundo—poco común en campos de fuerza aprendidos por máquina de vanguardia. Aún más notable, las simulaciones se ejecutaron de forma eficiente en CPUs estándar, paso a paso rivalizando o superando a algunos potenciales de redes neuronales prominentes que requieren GPUs potentes. En todo momento, las moléculas exploraron conjuntos ricos de geometrías y estados metaestables, mostrando que la robustez no se logró congelando artificialmente el movimiento ni imponiendo estructuras rígidas.
Qué significa esto para el modelado molecular futuro
Para quienes no son expertos, el mensaje clave es que no todos los modelos de aprendizaje automático con errores bajos son de fiar cuando se les exige mucho. Al anclar sus modelos en energías atómicas derivadas cuánticamente y al ajustar cuidadosamente las expectativas incorporadas del modelo hacia estados de alta energía, los autores han creado una familia de potenciales que generan de forma natural "fuerzas restauradoras"—el equivalente molecular de un arnés de seguridad—que mantienen las simulaciones dentro de la física incluso a altas temperaturas y desde puntos de partida distorsionados. Este enfoque promete simulaciones más fiables y prolongadas de moléculas complejas, y señala posibles extensiones futuras donde modelos informados por la física similares aborden fases condensadas e interacciones sutiles como la dispersión, todo ello manteniendo la practicidad computacional.
Cita: Isamura, B.K., Aten, O., Nosratjoo, M. et al. Unprecedented robustness of physics-informed atomic energy models at and beyond room temperature. Commun Chem 9, 138 (2026). https://doi.org/10.1038/s42004-026-01965-0
Palabras clave: campos de fuerza por aprendizaje automático, robustez de dinámica molecular, potenciales de procesos Gaussianos, modelado informado por la física, energías atómicas cuánticas