Clear Sky Science · es
Ajuste fino de AlphaFold con observaciones limitadas de cryo-EM
Por qué es tan difícil ver las formas de las proteínas
Las proteínas son diminutas máquinas moleculares que impulsan casi todos los procesos en nuestro organismo, desde la producción de energía hasta la transmisión de señales nerviosas. Para entender cómo funcionan —y cómo los fármacos podrían controlarlas— los científicos necesitan conocer sus formas tridimensionales precisas. Han surgido dos herramientas potentes para esta tarea: la crio‑microscopía electrónica (cryo‑EM), que toma muchas imágenes borrosas de proteínas congeladas, y AlphaFold, un sistema de inteligencia artificial que predice estructuras proteicas a partir de sus secuencias. Pero en muchos experimentos reales los datos de cryo‑EM son incompletos y las predicciones de AlphaFold no siempre coinciden con la realidad. Este artículo presenta CoCoFold, un método que enseña a AlphaFold a escuchar directamente datos de cryo‑EM difíciles y mejorar sus predicciones en consecuencia.
Cuando la cámara ve demasiado poco
La cryo‑EM funciona al congelar por choque las proteínas e imagenar enormes cantidades de partículas individuales desde muchas direcciones, para luego combinar esas imágenes en un mapa 3D. En la práctica, sin embargo, los investigadores a menudo no disponen de suficientes imágenes buenas. A veces la proteína aparece solo brevemente en un estado de alta energía, por lo que se capturan muy pocas partículas. En otros casos, las proteínas prefieren ciertas orientaciones sobre la superficie del hielo, de modo que faltan muchos ángulos de visión. Ambos problemas conducen a mapas borrosos e incompletos que son difíciles de traducir en modelos atómicos fiables. El software existente puede encajar las estructuras predichas por AlphaFold en esos mapas, pero su éxito depende en gran medida de disponer desde el principio de datos nítidos y de alta resolución.
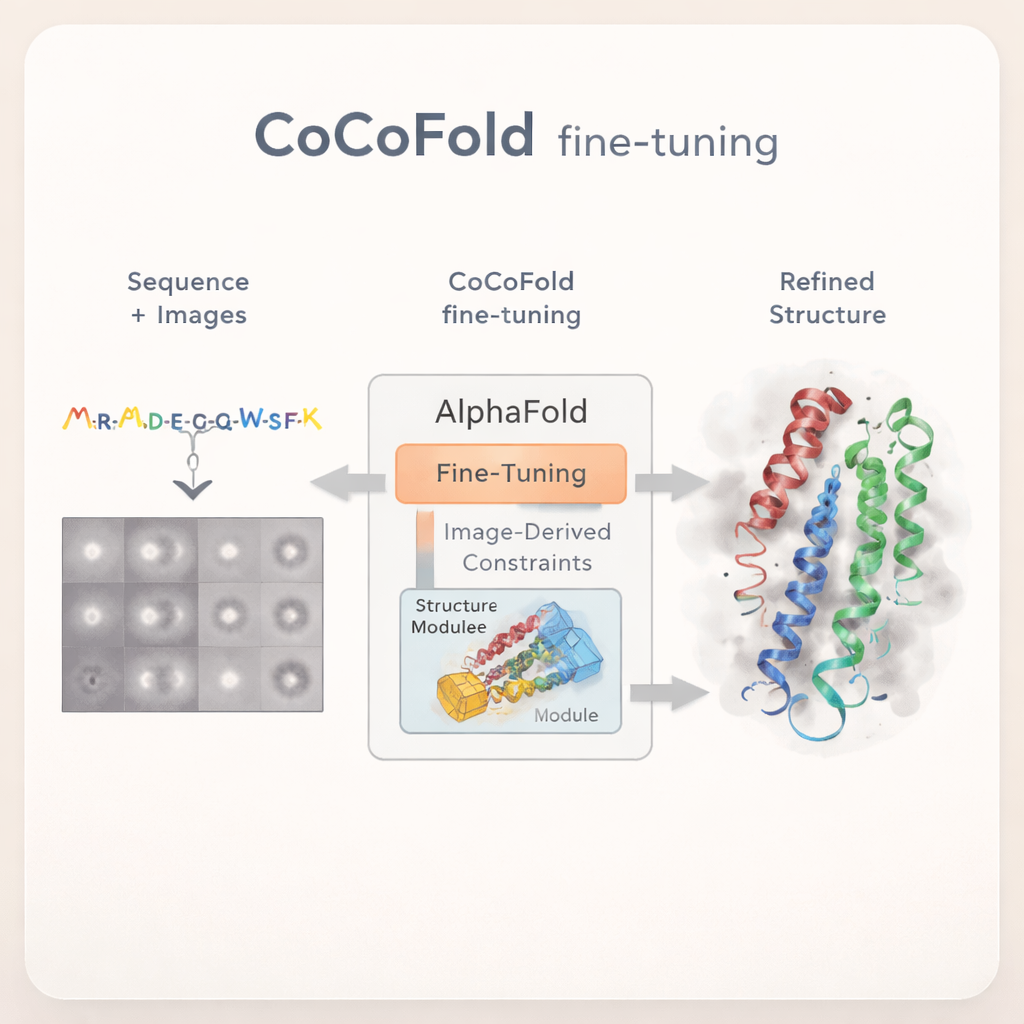
Enseñar a AlphaFold a aprender de imágenes crudas
CoCoFold adopta un enfoque diferente: en lugar de confiar en un mapa 3D de cryo‑EM totalmente reconstruido, utiliza directamente las imágenes 2D crudas de las partículas para afinar AlphaFold. El método parte de una predicción de AlphaFold‑Multimer y mantiene la mayor parte de la red original congelada, preservando su conocimiento amplio sobre el plegamiento proteico. Solo se permite cambiar la parte final que construye la estructura. Se añade un "adaptador" ligero para introducir información derivada de las imágenes de cryo‑EM en este módulo estructural, empujando suavemente el modelo hacia formas compatibles con los datos experimentales sin permitir desviaciones drásticas de la física conocida de las proteínas.
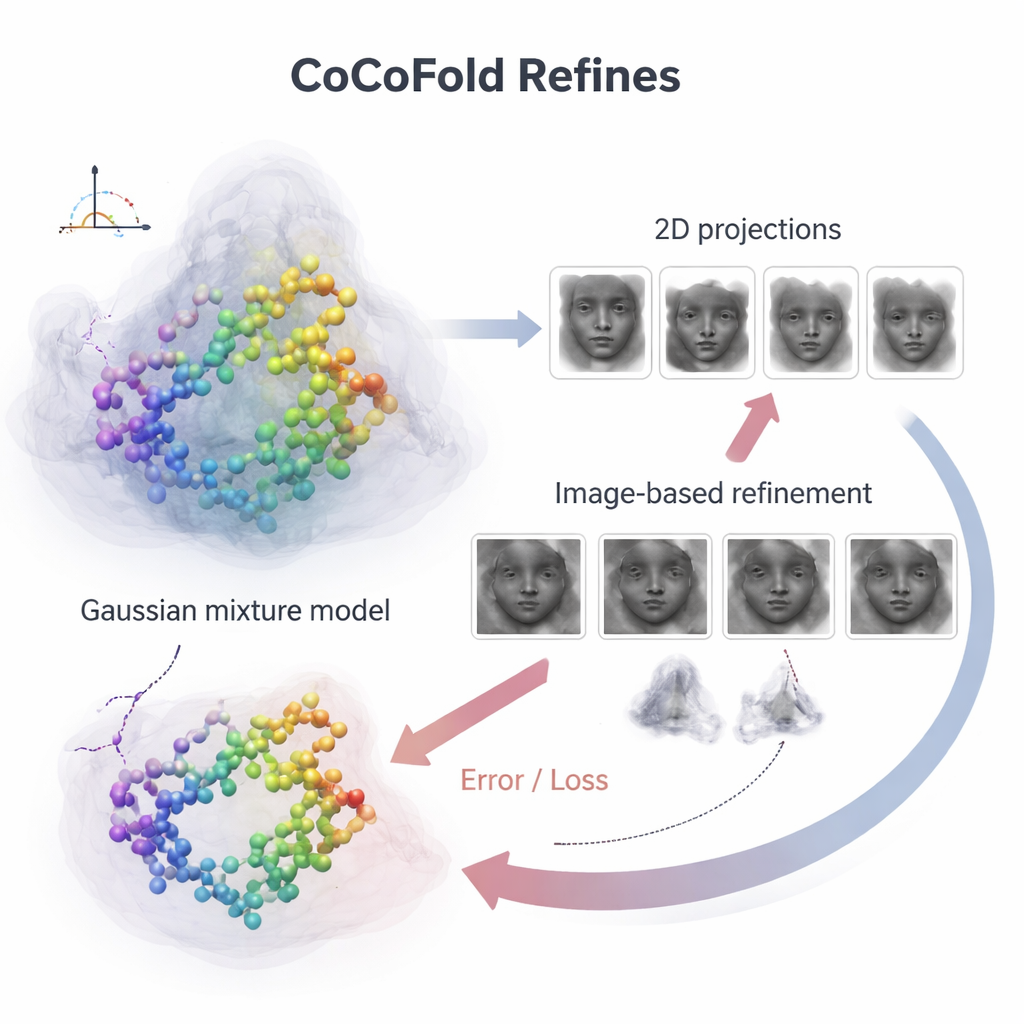
Convertir imágenes en retroalimentación estructural
Para conectar átomos individuales de la proteína con las ruidosas imágenes del microscopio, CoCoFold construye una representación suave y flexible de la estructura predicha usando blobs tridimensionales superpuestos, conocidos como mezcla gaussiana. A partir de esta representación simula cómo se vería la proteína en el microscopio con las mismas direcciones de visión y condiciones de imagen que el experimento real. Estas instantáneas simuladas se comparan luego con las partículas reales de cryo‑EM, anillo por anillo en el dominio de la frecuencia, para evaluar su concordancia. Cualquier desajuste se convierte en una señal de retroalimentación que fluye de vuelta a través de la red, ajustando ligeramente tanto el modelo proteico como la representación de densidad. Tras el entrenamiento, el modelo atómico se limpia adicionalmente mediante un paso de refinamiento basado en física para eliminar choques geométricos locales.
Mantener la precisión cuando los datos son escasos o sesgados
Los autores probaron CoCoFold en varios conjuntos de datos experimentales y simulados diseñados para imitar los dos problemas principales en cryo‑EM: muy pocas partículas y grandes huecos en los ángulos de visión. Bajo estas condiciones difíciles, las herramientas estándar —incluyendo otros métodos de aprendizaje profundo que dependen de mapas reconstruidos— tendían a perder regiones de la proteína, desplazar hélices o perder detalles finos a medida que los mapas se volvían más borrosos. CoCoFold, en contraste, produjo de forma constante modelos que coincidían más y mejor con las estructuras de referencia conocidas. Sus errores se mantuvieron pequeños incluso cuando el número de partículas se redujo drásticamente o cuando faltaban grandes conos de direcciones de visión, lo que sugiere que aprender directamente de las imágenes crudas conserva información crucial que los enfoques basados en mapas descartan.
Qué significa esto para la biología estructural futura
Para el público no especializado, el mensaje clave es que CoCoFold actúa como un traductor entre potentes predicciones de IA y datos experimentales imperfectos. En lugar de confiar únicamente en AlphaFold o en la cryo‑EM, permite que ambos se informen mutuamente, especialmente en los regímenes difíciles donde los experimentos ofrecen solo una vista parcial. En casos sencillos con datos abundantes y de alta calidad, las herramientas guiadas por mapas siguen funcionando muy bien. Pero cuando las partículas son raras o faltan orientaciones —situaciones comunes al seguir estados proteicos fugaces o frágiles— CoCoFold ofrece una manera de recuperar modelos atómicos fiables a partir de información que de otro modo se desperdiciaría.
Cita: Liao, J., Zheng, D., Zhang, H. et al. Fine-tuning AlphaFold with limited cryo-EM observations. Commun Chem 9, 95 (2026). https://doi.org/10.1038/s42004-026-01899-7
Palabras clave: cryo-EM, AlphaFold, estructura de proteínas, aprendizaje profundo, biología estructural