Clear Sky Science · es
El fallo energético mitocondrial subyace a la neuropatía sensorial relacionada con FLVCR1
Cuando los nervios del dolor se quedan sin energía
Algunas personas nacen casi sin capacidad para sentir dolor. A primera vista esto puede parecer una ventaja, pero pronto se convierte en una maldición: sin el dolor como señal de alarma sufren quemaduras, fracturas, infecciones e incluso ceguera. Este estudio investiga una forma poco común de esos trastornos de pérdida del dolor y descubre un culpable sorprendente: diminutas centrales eléctricas dentro de las células nerviosas cuya producción de energía falla de forma grave.
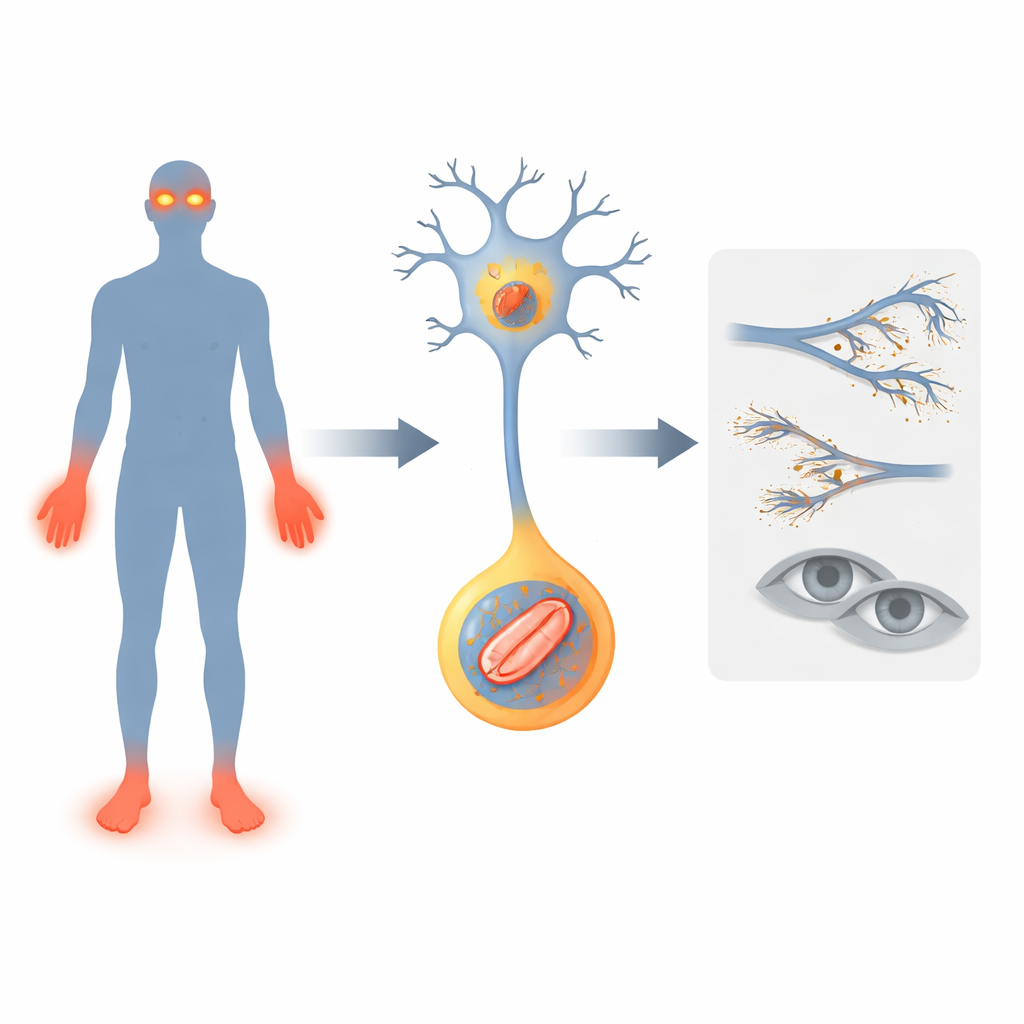
Un gen que apaga las campanas de alarma
Los investigadores se centran en un gen llamado FLVCR1, ya vinculado a enfermedades nerviosas raras en las que las personas pierden la sensación del dolor, desarrollan marcha inestable y, a veces, pérdida progresiva de la visión. Describen dos nuevos pacientes con variantes en FLVCR1. Ambos niños mostraron problemas tempranos: retraso en los hitos motores, caídas frecuentes, infecciones profundas y mutilación de dedos y dedos de los pies porque las lesiones pasaban desapercibidas. Uno además desarrolló una enfermedad ocular degenerativa llamada retinitis pigmentosa, que causa ceguera nocturna. Estos casos amplían el panorama de cómo las alteraciones de FLVCR1 pueden manifestarse en humanos y refuerzan la idea de que este gen es vital para mantener con vida a los nervios sensoriales y a las células fotosensibles de la retina.
Modelando la enfermedad en pececillos
Para explorar cómo FLVCR1 afecta a los nervios sensoriales en desarrollo, el equipo recurrió al pez cebra, cuyos embriones transparentes permiten ver directamente las células nerviosas. Redujeron los niveles de la versión del gen en pez, flvcr1a, usando herramientas genéticas. Los peces con flvcr1a disminuido tenían menos ganglios de la raíz dorsal, cúmulos de neuronas que detectan el tacto y el dolor a lo largo de la columna. A nivel de comportamiento, estos peces se movían menos espontáneamente y nadaban solo distancias cortas cuando les tocaban la cola suavemente, lo que sugiere una respuesta sensorial atenuada. Dado que modelos previos en ratón morían demasiado pronto para analizar sus nervios sensoriales, estos peces cebra proporcionan el primer sistema vivo en el que se pueden seguir en detalle los defectos nerviosos y el comportamiento relacionados con FLVCR1.
Tres vías alteradas convergen en las centrales celulares
FLVCR1 se localiza en membranas celulares y gestiona varias sustancias clave. Trabajos previos sugerían roles en el manejo de la colina (un componente para los lípidos de membrana), el hemo (el pigmento que contiene hierro que impulsa muchas enzimas) y el flujo de calcio entre compartimentos celulares. Los científicos recolectaron células de la piel (fibroblastos) de cuatro pacientes con distintas mutaciones en FLVCR1 y las compararon con células de personas sanas y portadores asintomáticos. Encontraron que las células de los pacientes tenían niveles más bajos de colina y membranas celulares más fluidas, cambios que podrían alterar el delicado entorno lipídico que requieren las mitocondrias, los orgánulos generadores de energía. También descubrieron que una enzima crucial para fabricar hemo dentro de las mitocondrias, ALAS1, estaba menos activa, aunque el contenido total de hemo parecía casi normal. Al mismo tiempo, los sitios de contacto físico entre el retículo endoplásmico y las mitocondrias —donde normalmente fluye calcio hacia las mitocondrias— eran más cortos y menos frecuentes, y la entrada de calcio en las mitocondrias estaba reducida. Tres problemas —escasez de colina, producción de hemo lenta y transferencia de calcio debilitada— apuntaban todos a un rendimiento mitocondrial deteriorado.
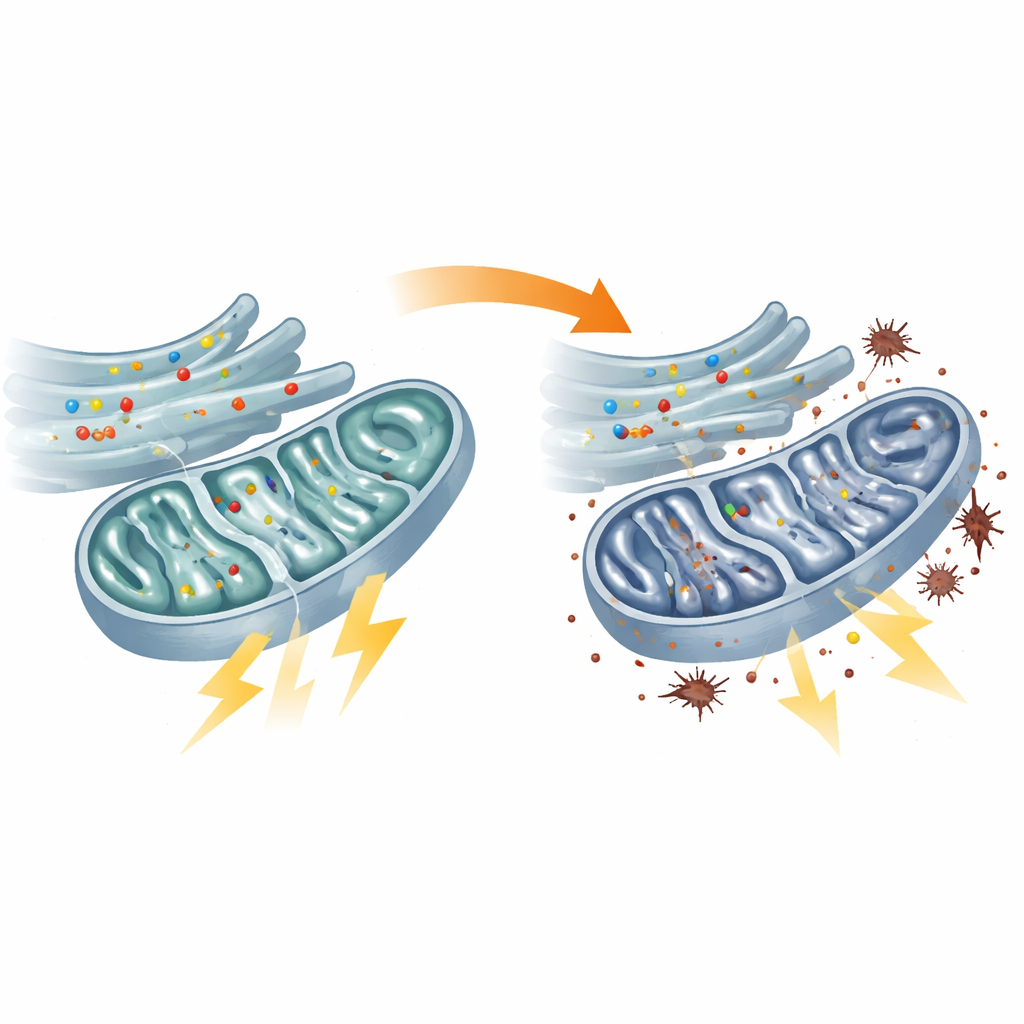
Mitocondrias hambrientas y sistemas de respaldo sobrecargados
Pruebas directas del metabolismo energético confirmaron que las mitocondrias en los fibroblastos de los pacientes funcionaban por debajo de lo normal. El eje central de procesamiento de combustible conocido como ciclo del TCA funcionaba más lentamente, varias de sus enzimas clave tenían menor actividad y la cadena de reacciones que normalmente convierte el combustible en ATP, la moneda energética de la célula, estaba deprimida. Como resultado, los niveles de ATP dentro de las mitocondrias disminuyeron. Las células intentaron compensar aumentando la glucólisis, una vía de quema de azúcar menos eficiente y localizada fuera de las mitocondrias. Este cambio de estrategia energética tuvo un coste: electrones se escaparon de la maquinaria mitocondrial estresada y desencadenaron niveles más altos de peroxidación lipídica, una forma de daño oxidativo a las membranas celulares. Defectos similares se observaron en peces cebra con flvcr1a reducido, vinculando directamente el fallo mitocondrial con el modelo animal de neuropatía sensorial.
Pistas para tratamientos futuros mediante el refuerzo energético celular
De forma alentadora, algunos de estos defectos pudieron mitigarse en el laboratorio. Cuando el equipo aumentó artificialmente la entrada de calcio en las mitocondrias sobreexpresando una proteína de canal llamada MCU en células de pacientes, la producción de energía se recuperó y los signos de daño oxidativo disminuyeron. Suministrar a las células un precursor del hemo, el ácido 5-aminolevulínico (ALA), también mejoró la actividad del ciclo del TCA, la función de la cadena respiratoria y los niveles de ATP, aunque exposiciones prolongadas a ALA resultaron dañinas en estudios previos. La colina extra normalizó la fluidez de la membrana y ayudó a reducir el daño lipídico, pero solo aportó ganancias modestas y a corto plazo en la producción de energía. Estos experimentos de rescate sugieren que ninguna vía por sí sola es la responsable; en cambio, una red de alteraciones en la gestión de colina, hemo y calcio empuja a las mitocondrias a un rendimiento crónicamente insuficiente.
Por qué estos hallazgos importan para los pacientes
Trazando las consecuencias de las mutaciones en FLVCR1 desde las moléculas a las células y a los organismos completos, este trabajo propone que el fallo energético mitocondrial es una fuerza motriz detrás de esta forma de neuropatía por pérdida del dolor y sus problemas visuales asociados. Los nervios sensoriales y los fotorreceptores tienen demandas energéticas inusualmente altas porque mantienen axones largos o renuevan continuamente las estructuras fotosensibles, lo que los hace especialmente vulnerables cuando la producción mitocondrial decae. El modelo en pez cebra y las células derivadas de pacientes ofrecen ahora plataformas prácticas para probar terapias que refuercen el metabolismo mitocondrial. Aunque tratamientos como la suplementación con colina, el impulso controlado del hemo o fármacos que aumenten la captación de calcio mitocondrial requerirán evaluación cuidadosa en modelos animales y ensayos clínicos, el mensaje central es claro: restaurar el suministro energético de neuronas frágiles podría algún día ayudar a proteger a las personas que nacen sin la señal de alarma más importante de la naturaleza: el dolor.
Cita: Bertino, F., Zanin Venturini, D.I., Grasso, E. et al. Mitochondrial energetic failure underlies FLVCR1-related sensory neuropathy. Commun Biol 9, 429 (2026). https://doi.org/10.1038/s42003-026-09691-y
Palabras clave: neuropatía sensorial, disfunción mitocondrial, FLVCR1, insensibilidad al dolor, metabolismo energético nervioso