Clear Sky Science · es
La desmetilasa de histonas KDM7A regula negativamente la polarización de macrófagos pro-fibróticos y la progresión de la fibrosis pulmonar
Por qué importan a todos los pulmones con cicatrices
Cuando los pulmones desarrollan cicatrices persistentes, respirar se convierte en una lucha cotidiana. Esta afección, conocida como fibrosis pulmonar, afecta a millones y hoy no tiene cura: solo existen fármacos que ralentizan el daño. En este estudio, los investigadores descubren un «freno» molecular hasta ahora oculto dentro de células inmunitarias llamadas macrófagos que ayuda a contener la formación de cicatrices en el pulmón. Comprender este freno podría abrir la puerta a nuevos tratamientos, no solo para la fibrosis pulmonar, sino potencialmente para otras enfermedades en las que la cicatrización dañina y la inflamación descontrolada van de la mano.
La historia de unas células inmunitarias camaleónicas
Los macrófagos son células inmunitarias de primera línea que patrullan los tejidos, eliminan restos y ayudan a reparar el daño. Pero también son camaleónicos: en algunas situaciones se convierten en combatientes proinflamatorios, mientras que en otras se transforman en reparadores que pueden fomentar la formación de cicatrices. Un tipo particular que promueve la cicatrización, llamado macrófagos profibróticos (Fib-Mac), está fuertemente ligado a la fibrosis pulmonar. Estas células producen moléculas que activan a los fibroblastos, que a su vez depositan colágeno y otros componentes de la matriz en exceso, endureciendo lentamente el pulmón. Los autores quisieron saber cómo las «configuraciones» genéticas dentro de los macrófagos determinan si se convierten en estas peligrosas células Fib-Mac o permanecen en estados más equilibrados y protectores.

Un freno epigenético escondido en el genoma
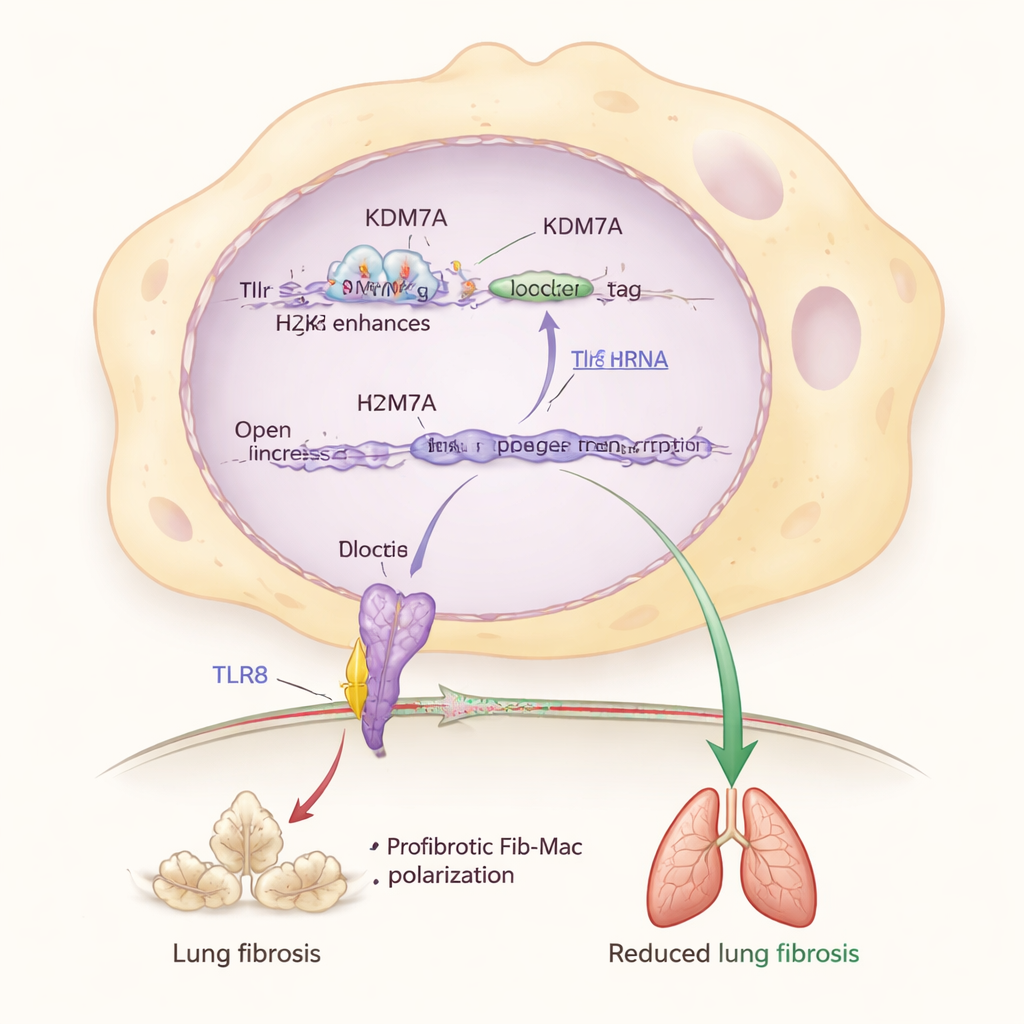
El equipo comenzó escaneando cientos de reguladores epigenéticos conocidos—proteínas que afinan cómo se empaqueta el ADN y qué genes se activan o silencian. Usando secuenciación de ARN en macrófagos humanos y de ratón, encontraron que una enzima llamada KDM7A se activaba fuertemente cuando los macrófagos se empujaban hacia un estado fibrótico y reparador. KDM7A es una «desmetilasa de histonas»: elimina ciertas marcas químicas de las proteínas histonas alrededor de las cuales se enrolla el ADN. Ese patrón sugería que KDM7A podría actuar como un freno de retroalimentación incorporado, activado precisamente cuando los macrófagos comienzan a desplazarse hacia una identidad promotora de cicatrices.
Para poner esto a prueba, los investigadores utilizaron ratones que carecen del gen Kdm7a e indujeron lesión pulmonar con el fármaco quimioterápico bleomicina, un modelo estándar de fibrosis pulmonar. Al principio tras la lesión, el tejido pulmonar se veía similar en animales normales y en los deficientes en Kdm7a. Pero a las tres semanas, los ratones sin Kdm7a mostraron cicatrices mucho más extensas, colapso de los alvéolos y puntuaciones de Ashcroft más altas que cuantifican la fibrosis. Los genes implicados en la producción de colágeno y otras vías relacionadas con la fibrosis estaban más activos en estos animales knockout, confirmando que la pérdida de Kdm7a hace que los pulmones sean más vulnerables a la formación de cicatrices duraderas y dañinas.
Cómo KDM7A aleja a los macrófagos de un destino pro-cicatriz
Usando secuenciación de ARN unicelular, los autores se centraron en células individuales del pulmón de ratones lesionados. Descubrieron que en ausencia de Kdm7a, un subconjunto particular de macrófagos en el tejido de sostén del pulmón se expandía de forma dramática y adquiría una fuerte firma Fib-Mac, expresando genes como Arg1, Spp1 y Trem2. Experimentos adicionales en macrófagos en cultivo mostraron que eliminar Kdm7a aumentaba los genes marcadores de Fib-Mac y reconducía el metabolismo celular hacia vías que apoyan la producción de colágeno y una activación sostenida. En otras palabras, KDM7A normalmente restringe tanto los programas genéticos como los metabólicos que empujan a los macrófagos hacia un estado promotor de fibrosis.
Ahondando más, los investigadores identificaron un socio clave en este sistema de freno: una proteína sensor llamada TLR8, que detecta fragmentos de ARN dentro de las células inmunitarias. Encontraron que KDM7A ayuda a mantener al gen Tlr8 activado al eliminar una marca química represiva (H3K27me2) de una región potenciadora cerca de Tlr8. Cuando Kdm7a fue desactivado, esta marca se acumuló, los niveles de Tlr8 cayeron y las características Fib-Mac se intensificaron. Reducir directamente Tlr8 en macrófagos también los empujó hacia una identidad fibrótica, mientras que activar o sobreexpresar TLR8 los devolvía atrás, incluso cuando faltaba Kdm7a. Esto sitúa la vía KDM7A–TLR8 en el centro de un circuito molecular que protege a los pulmones del exceso de cicatrización.
De pulmones envejecidos a la enfermedad humana
Para conectar estos hallazgos con las personas, el equipo examinó tejido pulmonar de pacientes con enfermedad pulmonar fibrótica. En comparación con tejido de control no enfermo, los pulmones fibróticos contenían muchos más macrófagos que portaban marcadores Fib-Mac, pero estas mismas células mostraban niveles notablemente reducidos de KDM7A y TLR8. Un nuevo análisis de conjuntos de datos unicelulares existentes de pacientes con fibrosis pulmonar idiopática confirmó este patrón: a medida que aumentaban las firmas Fib-Mac, la expresión de KDM7A disminuía. Los investigadores también exploraron un gran atlas de ratón y encontraron que la expresión de Kdm7a y Tlr8 en macrófagos disminuía con la edad en machos, reflejando el mayor riesgo de fibrosis pulmonar en hombres mayores. Esto sugiere que el debilitamiento relacionado con la edad y el sexo del freno KDM7A–TLR8 podría ayudar a explicar quiénes son más vulnerables a la cicatrización pulmonar grave.
Qué significa esto para tratamientos futuros
En términos sencillos, este trabajo muestra que nuestro sistema inmunitario lleva un mecanismo de seguridad interno que impide que las células reparadoras útiles se vuelvan demasiado entusiastas y se conviertan en impulsores de cicatrices permanentes. KDM7A, actuando a través de TLR8, evita que los macrófagos se bloqueen en un modo profibrótico y así ayuda a mantener un tejido pulmonar flexible y funcional tras una lesión. Cuando este sistema falla—por pérdida genética, envejecimiento u otros factores—los macrófagos tienen más probabilidades de convertirse en “amplificadores de cicatrices”, empeorando la fibrosis. Al revelar este freno epigenético, el estudio señala nuevas estrategias terapéuticas: fármacos que aumenten la actividad de KDM7A, imiten sus efectos o estimulen cuidadosamente TLR8 podrían algún día complementar las terapias antifibróticas existentes y ofrecer mejor protección frente a la cicatrización pulmonar progresiva y limitante para la vida.
Cita: Funagura, N., Koga, T., Etoh, K. et al. Histone demethylase KDM7A negatively regulates fibrotic macrophage polarization and lung fibrosis progression. Commun Biol 9, 309 (2026). https://doi.org/10.1038/s42003-026-09610-1
Palabras clave: fibrosis pulmonar, macrófagos, epigenética, KDM7A, TLR8